
#NephMadness 2019: Complement Region
Submit your picks! | NephMadness 2019 | #NephMadness | #ComplementRegion
Selection Committee Member: Carla Nester
Carla Nester is the Director of Pediatric Nephrology in the Stead Family Children’s Hospital at the University of Iowa and the Associate Director of the Molecular Otolaryngology and Renal Research Laboratory. Her research focuses on defining the role of complement dysregulation in the complement-mediated kidney diseases (particularly C3 Glomerulopathy).
Writer: Anuja Java @anuja_java
Anuja Java is an Assistant Professor in the Division of Nephrology at Washington University School of Medicine in St. Louis, MO. Her research focuses on defining the functional consequences of genetic mutations identified in complement-mediated kidney diseases such as atypical hemolytic uremic syndrome and C3 glomerulopathy.
Competitors for the Complement Region
Genetic C3GN vs Acquired C3GN
Eculizumab vs C3-i and Beyond

Copyright: Ernst Christen / Shutterstock
This NephMadness region features some of the oldest players in this year’s tournament. The early elements of the complement system appeared more than a billion years ago as the main player in innate immunity with the keen ability to self-amplify and coat pathogens. The system has retained its ancient primary function of opsonization in addition to developing features that enable it to mount a rapid, highly targeted, and powerful defense against microbes. The contemporary complement system thus lies at the interface of innate and adaptive immunity.
There are three major pathways of complement activation: classical, lectin, and alternative. The classical pathway (CP) is primarily triggered by antibodies binding to an antigen. In an analogous process, the lectin pathway (LP) is activated by lectin (equivalent to an antibody) binding to a sugar (equivalent to an antigen) on a microbe. Unlike the CP and LP, the alternative pathway (AP) has the capacity to deposit on a pathogen without need for prior contact/exposure. A key feature and common goal of the three pathways is C3 activation to generate C3b. The full cascade can be seen below:
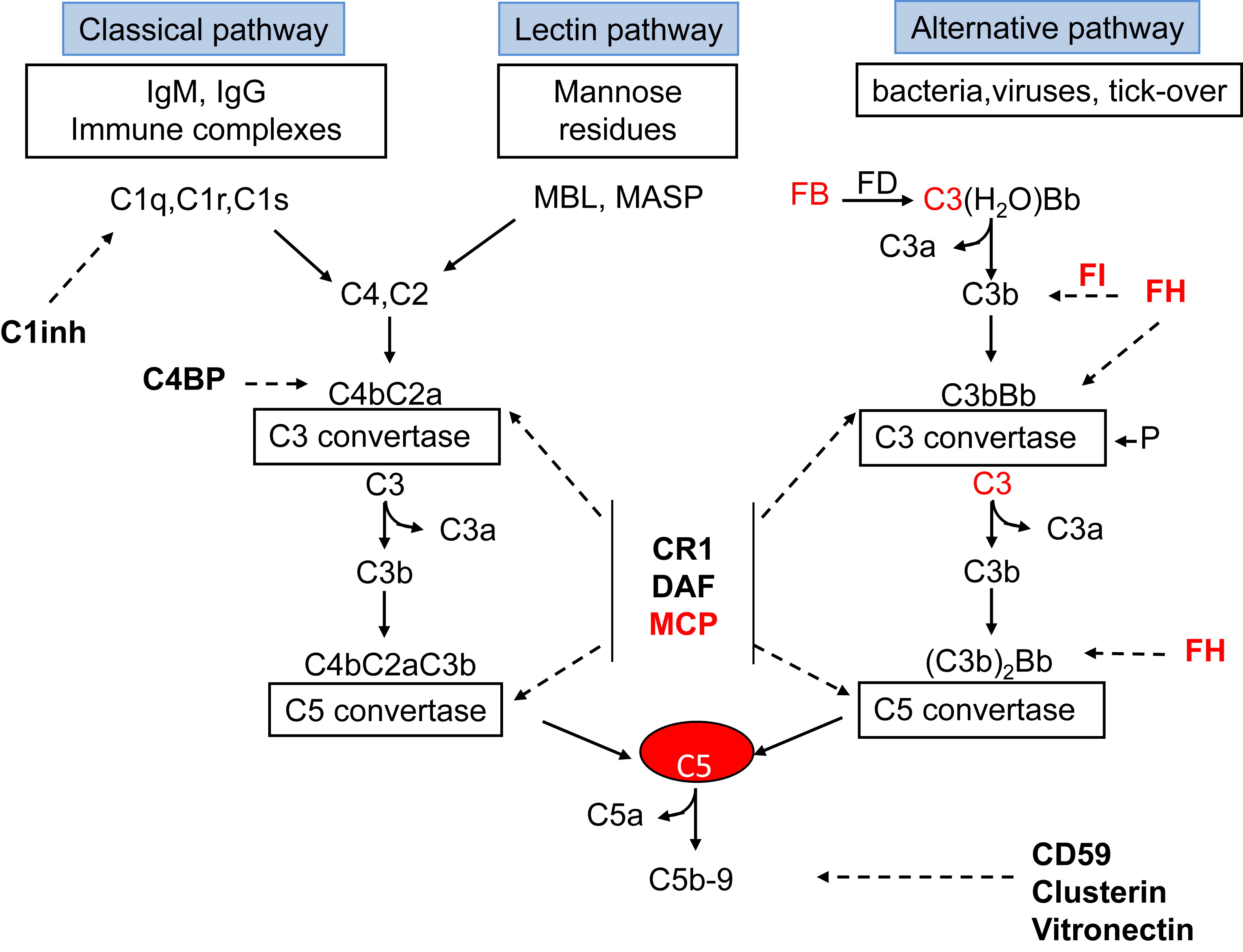
Fig 1 from Noris & Remuzzi, AJKD. © National Kidney Foundation.
After the initial recognition event, the CP and LP feature multiple proteolytic cleavage steps leading to activation of the components C2 and C4 and the formation of the CP C3 convertase (C4b2ba). This enzymatic complex converts C3 into C3a and C3b. The AP however, is continuously generating a small amount of auto-activated C3 (the so-called C3 tickover) via the spontaneous hydrolysis of C3. If this auto-activated C3 deposits on healthy self-tissue, host regulatory proteins immediately inactivate it. However, if it deposits on a microbe, activated C3 can be quickly amplified by engaging two proteases, factor B (FB) and factor D (FD), to create the AP C3 convertase (C3bBb).
Similar to the CP C3 convertase, this enzyme also converts C3 into C3a and C3b. The larger fragment (C3b) binds covalently to the target to mediate opsonization, and the smaller peptide (C3a) elicits a local inflammatory response. A critical feature of the AP is its amplification loop and C3b, no matter how it is generated, serves as a nidus for this feedback loop for the generation of large amounts of C3b to opsonize a pathogen. Addition of C3b to a C3 convertase generates a C5 convertase (C4b2aC3b or C3bBbC3b) that cleaves the C5 protein into C5a (a potent anaphylatoxin) and C5b [engages the membrane attack complex (MAC)]. Original figure available under a CC BY-NC-ND license.
Because the complement system provides a rapidly activated and potent surveillance mechanism designed to eliminate the pathogen, strict control is required to avoid damage to self. The regulatory proteins that protect against undesirable complement activation include two in plasma, factor H (FH) and factor I (FI), and three membrane proteins, namely, membrane cofactor protein (MCP; encoded by CD46), decay-accelerating factor (DAF; CD55), and complement receptor 1 (CR1, aka CD35).
Control at the critical central step of C3b generation is provided by two main mechanisms. One, called cofactor activity, refers to proteolytic cleavage of C3b and C4b. In this process, a cofactor protein (such as FH or CD46) binds to those fragments, then the plasma serine protease FI can proteolytically inactivate them. The second regulatory mechanism is called decay-accelerating activity whereby the C3 or C5 convertase is disassociated. Both mechanisms prevent activation of the powerful feedback loop on self. Thus, inadequate control of complement activation due to inherited defects in complement genes and acquired auto-antibodies against a regulatory protein can predispose to several diseases.
C3 glomerulopathy (C3G) is a glomerulonephritis that occurs secondary to uncontrolled activation of the alternative complement pathway leading to isolated or dominant C3 staining seen on immunofluorescence. The term C3G encompasses dense deposit disease (DDD, formerly known as membranoproliferative glomerulonephritis [MPGN] type 2) and C3 glomerulonephritis (C3GN; formerly known as MPGN type 1 or type 3). The two subtypes are differentiated based on the location of deposits on electron microscopy. DDD is characterized by electron-dense material in the glomerular basement membrane (GBM) while C3GN is characterized by subendothelial, mesangial, and in some cases, subepithelial deposits.
Excessive activation of the AP can occur from genetic or acquired factors as discussed further below. Recent reports have suggested that postinfectious GN (PIGN) and C3G may be two ends of a spectrum of the same glomerular disease. The prognosis of PIGN is typically good. However, if a patient with PIGN presents with atypical features, such as persistent proteinuria, low C3, and nonrecovery of kidney function, C3G should be suspected.
Now that we are all have a good understanding of the complement system and C3GN, let’s get to the scouting report for these four strong teams: Genetic C3GN, Acquired C3GN, Eculizumab, and C3 Inhibition!
Genetic C3GN

Copyright: Sergey Nivens / Shutterstock
The genetic drivers of disease include either loss-of-function mutations in regulators (FH, FI, or MCP) or gain-of-function mutations in activators [complement component 3 (C3) or factor B (FB)] of the AP. The genetic variants associated with C3G include rare variants that are either associated with familial disease (thus providing strong evidence that they are causative) or have been identified in individual patients and are thought to be likely pathogenic. Common polymorphisms that may increase the risk of disease in association with other genetic variants or environmental factors have also been reported.
Studies of familial C3G have revealed families with genetic deficiency of FH or with a mutation in FH that renders it less able to regulate the alternative pathway. In one report with 16 FH-deficient patients, four presented with MPGN and the other 12 developed atypical hemolytic uremic syndrome (aHUS). Additionally, a family with a variant in C3 has been described whereby the protein encoded by the C3 variant formed a C3 convertase resistant to decay by FH. Similarly, another pedigree showed a mutation of C3 leading to defective regulation by CR1 and FH. More recently, the role of FH-related proteins (CFHRs) has been implicated in C3G. There are five human CFHRs that are coded downstream of FH on chromosome 1. They are highly homologous to FH, especially at their carboxy-terminal ends. Five separate abnormal FHR proteins have been associated with C3G.
In addition to familial cases, other series have described sporadic rare genetic variants associated with C3G in individual patients. These include variants in genes coding for FH, FI, FB, MCP, C3, and thrombomodulin. Known common variants of complement genes that increase the risk of C3G include variants in FH and MCP.
Acquired C3GN
Acquired factors associated with C3 glomerulopathy include C3 nephritic factors (C3Nefs); autoantibodies to FH, FB, or C3b; as well as the presence of monoclonal gammopathy.
Copyright: Sakurra / Shutterstock
Of the acquired factors, the most common and the best studied are C3Nefs, reported in up to 80% of patients with DDD and 50% of patients with C3GN. C3Nefs are autoantibodies that bind to and stabilize the AP C3 convertase (C3bBb) by preventing its inactivation by FH. This leads to a prolonged C3 convertase activity, resulting in continued conversion of C3 to C3b, which in turn causes consumption of C3. However, C3Nefs are heterogeneous and there is a lack of standardized assays for their measurement, making comparisons between different cohorts difficult. In rare patients, autoantibodies to FH, FB, or C3b have also been identified.
Additionally, the association between monoclonal gammopathy and C3G has been clearly established by several reports. In one study, 65% of patients with C3G who were over 50 years of age also had a monoclonal gammopathy present. This was 16 times higher than the prevalence of monoclonal gammopathy in the general population. C3Nef was identified in 45% of these patients, suggesting that the monoclonal immunoglobulin may act as an autoantibody to C3 convertase or to other complement components. Targeted treatment of the monoclonal immunoglobulin resulted in complete or partial renal response or stable kidney function in 43.8% of these patients.

Visual Abstract by @beans_sam on Ravindran et al
These findings emphasize the importance of testing for monoclonal immunoglobulin in older patients with C3 glomerulopathy. Of note, targeted treatment was given preferentially to patients who had multiple myeloma, whereas patients with monoclonal gammopathy of renal significance (MGRS) received nontargeted treatment. However, of the patients achieving a hematologic response, 70% of patients achieved better renal outcomes. As the hematologic response was associated with good renal response, Ravindran et al suggest that targeted therapy should also be considered for patients with MGRS.
Eculizumab
Eculizumab is a humanized monoclonal antibody directed against the terminal pathway protein C5 (part of the membrane attack complex [MAC]) as seen in the figure below. It binds to and blocks the amino-terminal cleavage of C5 into C5b and C5a, thereby preventing formation of the MAC, C5b-9, and the liberation of C5a. This prevents the proinflammatory effects of C5a and the cytolytic effects of the MAC. Because it acts downstream of C3, eculizumab preserves the autoimmune protective and immune-enhancing functions of the opsonin C3b and the anaphylatoxin C3a.

Involvement of complement system in thrombotic microangiopathy (TMA) and eculizumab treatment. Figure 1 from Schmidtko et al, AJKD, © National Kidney Foundation.
Eculizumab was first approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) in 2007. In September 2011, it was approved by the FDA to treat aHUS based on two prospective studies in a total of 37 patients, along with a retrospective study of 19 patients. These studies demonstrated the efficacy and safety of eculizumab for treating aHUS in patients who were either unresponsive to or dependent on plasma exchange. These initial trials demonstrated that eculizumab was associated with improvement in kidney function in approximately 85% of aHUS patients. The two-year follow-up analysis established that the early clinical benefits achieved by eculizumab were maintained at two years. Recent evidence suggests that earlier initiation of treatment leads to improved results in terms of kidney function.

Visual Abstract by @divyaa24 on Legendre et al
Patients with aHUS who progress to kidney failure are candidates for kidney transplantation. However, there is a >50% risk of aHUS recurrence, with graft loss occurring in 80%-90% of the cases. The outcome, though, can vary depending on the complement abnormality. The risk of recurrence is 70%-90% with a FH mutation, 50%-80% with a FI mutation, and 40%-70% with a C3 mutation. The recurrence rate is low in patients with MCP mutations because the allograft expresses normal membrane-bound MCP. Data from 2014 show that eculizumab is also effective for the treatment of aHUS recurrence after kidney transplantation and as prophylaxis before transplantation in patients with a known pathogenic mutation. Eculizumab is now the standard of care for PNH and aHUS. This monoclonal antibody therapy should be initiated early to offer the best chance to recover kidney function.
Similar to aHUS, complement activation also plays a role in the pathogenesis of C3G. The pathophysiology of C3G suggests that a C3 convertase inhibitor, which would limit C3 breakdown product deposition on basement membranes, would be a strong therapeutic option. Although trials are ongoing for some novel targets, there is no FDA-approved drug for treatment of C3G that specifically inhibits the early components of the AP required for C3 convertase. Given this gap, eculizumab has been tried in patients with C3G, but the results (mostly in the form of case reports and one small prospective trial) have been mixed. These limited data suggest that eculizumab is effective in approximately 50% of patients with C3G and that it is most efficient in treating rapidly progressive and crescentic forms of C3G. Formal prospective studies are needed to assess the effect of eculizumab as a therapeutic agent in C3G.

Copyright: Brian A Jackson / Shutterstock
Dose: Eculizumab is administered intravenously and has a half-life of approximately 11 days. The recommended dosage regimen is 900 mg weekly for four doses, then 1,200 mg at week five, followed by 1,200 mg every two weeks. In patients receiving plasmapheresis, a supplemental dose is administered after each plasma exchange.
Trials are underway for a long-acting C5 inhibitor that can be administered every 8 weeks in patients with aHUS and PNH.
Duration of treatment: Eculizumab is well tolerated and relatively safe. The optimal duration of therapy is unclear. Eculizumab has been one of the most expensive drugs on the market since its release. The almost prohibitive cost of this drug and other issues mean that the duration of treatment should be tailored based in part at least on individual complement genetics. Lifelong treatment may be needed in patients with pathogenic mutations known to be associated with poor outcomes (for example, FH loss-of-function or C3 gain-of-function mutations), whereas withdrawal of eculizumab is more likely to be successful in patients with isolated MCP mutations.
The duration of treatment in patients with a mutation of unknown functional consequence and in those with no identified genetic abnormalities remains a largely unexplored issue. These remaining questions underscore the necessity to continue therapeutic studies, in particular to employ functional characterization of genetic variants of uncertain significance and further to conduct larger-scale, prospective randomized trials. There is much yet to learn about the duration, frequency, and magnitude of treatment with eculizumab.
Adverse effects: The most frequently reported adverse reactions with eculizumab include hypertension, headache, upper respiratory tract infection, urinary tract infection, nausea, vomiting, diarrhea, anemia, and leukopenia. The relationship, if any, of these relative minor side effects to drug activity is unclear. The predominant concern is a life-threatening infection with N. meningitidis (due to blockage of the terminal complement pathway). A meningococcal vaccination must be administered to everyone undergoing treatment with eculizumab.
Physicians need to be aware that the standard vaccine does not protect against the B serotype of the bacteria nor many of the infrequent (rare) serotypes. The Centers for Disease Control and Prevention now recommend serotype B Neisseria vaccine in patients receiving eculizumab as well as the standard Neisseria vaccine. In addition, appropriate antibiotics should be used for at least 14 days if there is not enough time to wait for the immune response. Moreover, suggestive symptoms of a bacteremia or septicemia should necessitate urgent investigation and antibiotic therapy.
C3 Inhibition (And Beyond)
Novel drug development for complement-mediated diseases has focused on new therapeutic options on all three pathways of complement activation (classical, lectin, and alternative), some targeting steps early in the complement cascade and others intervening downstream.

Regulation of the C3b alternative pathway amplification or feedback loop. Adapted from Figure 3: Java et al, Annual Review of Medicine
Some of the drugs in development are described below:
- Factor D inhibitor (ACH-4471): Factor D is a highly specific serine protease that cleaves Factor B into Ba and Bb. The fragment Bb combines with C3b to form the powerful alternative pathway C3 convertase (C3bBb), which can generate large amounts of C3b via the amplification loop. An orally available inhibitor of Factor D has been developed by Achillion Pharmaceuticals for C3G. A trial of 14-day treatment period initiated in September 2017 on four patients showed >50% reduction in proteinuria as measured by the albumin-creatinine ratio (ACR). Additionally there was a 30%-50% reduction in levels of Bb. The drug was well tolerated by the patients and is currently being tested in 6-month and 12-month open-label studies.
- C3 Inhibitor (AMY-101): Cp40 is a second-generation compstatin analog developed by Amyndas Pharmaceuticals. Compstatins are cyclic peptides that inhibit complement activation by binding C3 and interfering with convertase formation and thereby C3 cleavage. The rationale behind developing this drug was that targeting C3 would provide an effective treatment for C3G. Cp40 was investigated in vitro and was found to prevent complement-mediated lysis of sheep red blood cells in sera from C3G patients and arrested abnormal C3 turnover by directly inhibiting the nephritic factors (C3Nef and C4Nef). Cp40 has been granted an orphan drug status by the US FDA and the European Medicines Agency (EMA) for C3G. Clinical trials are ongoing.

Copyright: Gorodenkoff / Shutterstock
- C5a Receptor (C5aR) Inhibitor (avacopan; CCX168): Avacopan is an orally administered small-molecule inhibitor of the C5aR and thus blocks the activity of complement C5a. This drug, developed by ChemoCentryx, protected mice in a model of ANCA-associated vasculitis (AAV). Enrollment for a phase 3 clinical trial for AAV has recently been completed. Preliminary results from phase 2 trials indicated that the drug improves overall disease activity, including renal manifestations. The company has an ongoing phase 2 proof-of-concept clinical trial in patients with aHUS who are on dialysis. The primary efficacy objective of the trial is to evaluate whether treatment with avacopan may reduce thrombosis formation in maintenance dialysis patients with aHUS. The drug is being dosed under a compassionate use protocol.The drug has also been reported to be used in a kidney transplant recipient with C3G. Prior to being treated with avacopan, the patient had deteriorating kidney function and had been unsuccessfully treated with a wide spectrum of immunosuppressant drugs. After one month of treatment with avacopan, the patient’s kidney function (based on estimated glomerular filtration rate) stabilized. Sequential kidney biopsies taken 2 and 7 months post-treatment showed continued improvement in kidney histology. Enrollment is currently ongoing for a clinical trial of avacopan in C3G, the primary end point being the percentage change in the C3 histological index after six months of treatment. The drug has now been given orphan drug designation by the FDA for AAV, C3G, and aHUS.
- Mannan-binding lectin-associated serine protease 2 (MASP-2) Inhibitor (OMS721): MASP-2 is an effector enzyme required for the function of the lectin pathway and is thus a novel proinflammatory target. OMS721, a fully human monoclonal antibody targeting MASP-2, has been developed by Omeros for hematopoietic stem-cell transplant−related thrombotic microangiopathy, aHUS, and IgA nephropathy. Phase 2 and 3 clinical trials are in progress in these diseases.
Future Directions
The kidney is very susceptible to complement-mediated injury and thus the development of novel complement inhibitory drugs is critical to improve care in many different kidney diseases. Clinical trials of eculizumab have demonstrated the efficacy of complement blockade in aHUS. Better biomarkers of complement activation will be useful for rapidly confirming the biological effects of the new drugs. As novel therapies become available, however, a major challenge will be to design multicenter clinical trials that can adequately test these numerous different agents in patients with rare or slowly progressive kidney diseases.
– Executive Team Member for this region: Anna Burgner, AJKD Social Media Advisory Board member. Follow her @anna_burgner.
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC point for reading this region.
- Register/log in to the NKF’s Professional Education Resource Center (PERC).
- Review the activity and accreditation information.
- Click “Continue” and review Course Instructions.
- Complete Post-Test. Please note: By selecting “Yes” to the participation questions for each region, the corresponding Post-Test questions will appear. Click “Save Draft” to save your responses and finish later. When you are ready to submit your answers, click “Preview” to review all responses, then click “Submit.”
- Click “Next” to complete the Evaluation form, then click“Submit.”
- Claim 1.0 CME credit and 1.0 MOC point per region (up to 8.0 total for 8 regions of NephMadness).
- Print your certificate.
- Review the Post-Test answers and rationale.
The CME and MOC activity will expire on June 15th, 2019.
Submit your picks! | #NephMadness | @NephMadness | #ComplementRegion
Leave a Reply