
Silencing Primary Hyperoxaluria with Lumasiran: Two Year Follow-up of the Phase 3 ILLUMINATE-C Trial
Matthew A. Sparks (@Nephro_Sparks or @nephrosparks.bsky.social) is an associate professor of medicine at Duke University in Durham, NC. He is the director of the nephrology fellowship program and lead of the Society for Early Education Scholars (SEEDS) program in the department of medicine. He has served as an unpaid consultant for Alnylam Pharmaceuticals, the manufacturer of lumasiran, regarding their drug zilebesiran.
Alessandra Tomasi (@AleTomasiMD) is a second year nephrology fellow at Duke University in Durham, NC. She completed her medical education at the University of North Carolina at Chapel Hill School of Medicine, and her internal medicine residency as well as a chief resident year at Mayo Clinic in Rochester, MN. She plans to pursue a career in academic nephrology, with a focus on glomerular disease and medical education.
Disease-modifying therapies aim to slow or even reverse disease progression by directly targeting their underlying causes. The concept of disease modification first came to fruition in the 1970s, initially with respect to therapies for rheumatoid arthritis, and over the following decades has since gained significant traction across various conditions.
The use of RNA interference describes an approach to disease-modifying therapy that harnesses non-coding RNA molecules to directly interfere with the expression of specific genes. First described in the 1990s, this tool ultimately causes specific and efficient gene silencing at the post-transcriptional stage, by preventing translation. Since the early 2000s, this technology has been increasingly used in drug development, including in clinical trials.
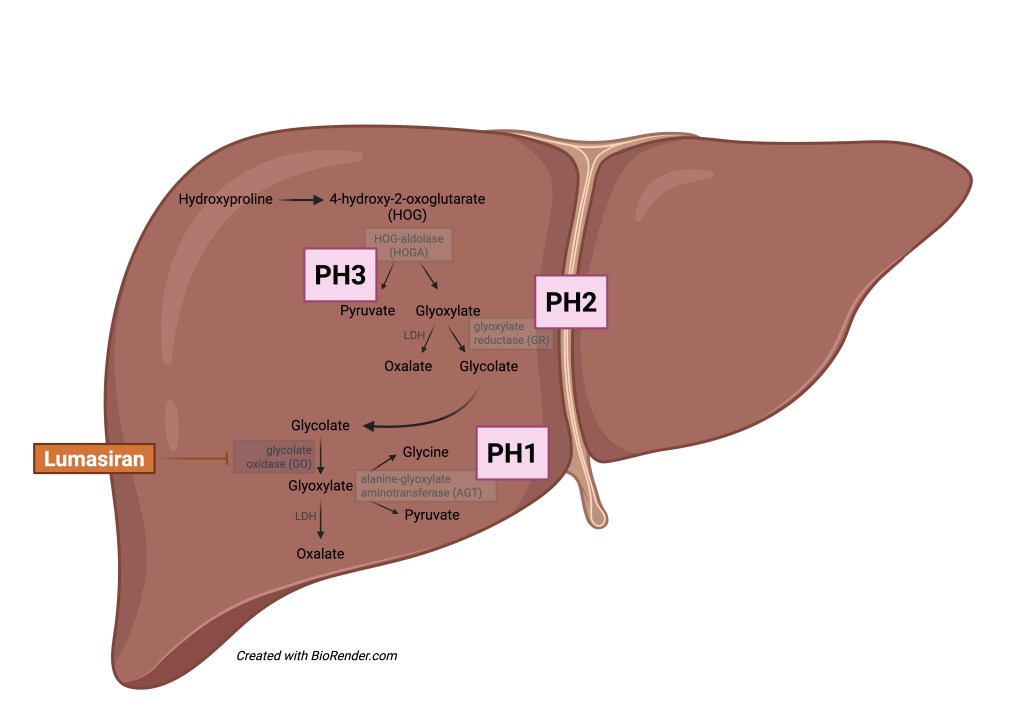
In the case of Primary Hyperoxaluria Type 1 (PH1), hepatic alanine-glyoxylate aminotransferase (AGT) deficiency leads to oxalate over-production, resulting in renal as well as systemic accumulation of calcium oxalate. This pathophysiology differs from that underlying Primary Hyperoxaluria Types 2 and 3 (PH2, PH3) which are caused by defects in glyoxylate reductase (GR) and 4-hydroxy-2-oxoglutarate aldolase (HOGA), respectively. Both PH2 and PH3 are less aggressive than PH1, as patients with PH2 generally have a slower progression of kidney disease and patients with PH3 very rarely progress to kidney failure.
© Tomasi
In the case of PH1, which is inherited in an autosomal recessive pattern, deficiency of the liver enzyme AGT as described above leads to increased oxalate production, resulting in nephrocalcinosis, nephrolithiasis, and chronic kidney disease. Management of PH1 is particularly challenging, and curative treatment ultimately culminates in the need for combined liver-kidney transplant.
In 2021, the ILLUMINATE -A trial demonstrated that lumasiran, an RNA interference (RNAi) therapeutic agent, reduces oxalate excretion to the point of normal or near-normal levels after 6 months of treatment in patients with PH1. It does so by silencing the gene that encodes glycolate oxidase, a key step in the pathway of glyoxylate (and ultimately oxalate) production. This study was followed by ILLUMINATE -C, which in 2023 demonstrated that lumasiran resulted in reduced levels of plasma oxalate levels in patients with PH1 who have kidney disease or were on dialysis already.
Now, 24 months later, all 21 patients who were enrolled in the ILLUMINATE -C trial were entered in the study’s extension period which was recently published in AJKD as a research letter. The authors show that at 2 years, reductions in plasma oxalate levels were sustained with continued use of lumasiran; additionally, its safety profile in both adults and children with PH1 were acceptable including in patients with advanced kidney disease.
Specifically, this study followed both pediatric and adult patients aged 4 to 59 years, who were assigned a cohort based on whether or not they were on hemodialysis. Subcutaneous lumasiran was administered at intervals based on body weight, and notably the majority of patients had evidence of systemic oxalosis, whether cardiac, skeletal, or ocular, at the time of enrollment. Among patients not yet on hemodialysis, mean plasma oxalate levels at 24 months decreased by 60% from baseline, and among those on dialysis, levels decreased by 30%. Of note, two patients underwent kidney-only transplant, and both continued lumasiran post-transplant with functioning grafts and minimal changes in eGFR at the end of the 2-year follow up period (two other patients withdrew from the study following combined liver-kidney transplant). Overall, lumasiran was well-tolerated, with the most frequently reported adverse effects being fever, diarrhea, and injection site reaction and no severe effects noted.
These data provide hope for new medical therapies to target PH1 and adds a significant advancement to the landscape of PH1 treatment. The question now remains whether reduced oxalate levels further translates into slowed progression to dialysis initiation and kidney/ liver transplantation in affected patients. Though we may never have a trial that randomizes people on and off therapy given the above findings, further research will continue to identify the long-term benefits of lumasiran, as well as optimal timing of therapy initiation and any potential role for combined therapy in the treatment of PH1.
-Post prepared by Matthew A. Sparks and Alessandra Tomasi
To view Sellier-Leclerc et al [FREE], please visit AJKD.org:
Title: Efficacy and Safety of Lumasiran for Advanced Primary Hyperoxaluria Type 1: 24-Month Follow-up of the Phase 3 ILLUMINATE-C Trial
Authors: Anne-Laure Sellier-Leclerc, Daniella Magen, Hadas Shasha-Lavsky, Eva Simkova, Arnaud Devresse, Fitsum Guebre-Egziabher, Mini Michael, John C. Lieske, Yaacov Frishberg, Sevcan A. Bakkaloglu, Chebl Mourani, Rola Saqan, Richard Singer, Isabella Guzzo, Nune Makarova, Richard Willey, Cristin Kaspar, John M. Gansner, and Jaap W. Groothoff
DOI: 10.1053/j.ajkd.2025.01.016
Leave a Reply