
#NephMadness 2026: Genetics Region
Submit your picks! | @NephMadness | @nephmadness.bsky.social | NephMadness 2026
Selection Committee Member: Eric Wallace
Eric Wallace is a Professor of Medicine in the Division of Nephrology at the University of Alabama at Birmingham. He is the Director of the Rare Genetic Kidney Disease. He is also the Chief Medical Information and Digital Health Officer of the UAB Health System which stemmed from his passion for the use of technology to expand access to care to those in need.
Writer: Steven Clapp
Steven Clapp is a Nephrology Fellow at Vanderbilt-University Medical Center in Nashville, Tennessee. He completed his residency in Internal Medicine and Pediatrics at the Indiana University School of Medicine. His interests include caring for individuals with genetic kidney disease, as well as transitions of care for children and young adults with chronic kidney disease.
Writer: Alyssa Steitz
Alyssa Steitz is a second-year nephrology fellow at Vanderbilt. She earned her medical degree from Florida State University and a master’s in education from Vanderbilt, where she also completed internal medicine residency and served as Chief Resident. Her interests include professional identity formation, cardiorenal syndrome, and palliative kidney care.



Competitors for the Genetics Region
There has been a recent explosion in the field of genetic nephrology. This has led to better diagnosis of the cause of patients kidney disease as well as many recent advancements in precision medicine to treat the genetic cause of kidney disease. In this region we focus on the growing knowledge surrounding cystic kidney diseases and then will dive into the growing options for treatment of Fabry Disease.
Team 1: PKD Masqueraders
versus
Team 2: Fabry Treatment
Image generated by Matthew Sparks using ChatGPT at http://chat.openai.com, February 2026. After using the tool to generate the image, Sparks and the NephMadness Executive Team reviewed and take full responsibility for the final graphic image.
Team 1: PKD Masqueraders
“‘When I see a bird that walks like a duck and swims like a duck and quacks like a duck, I call that bird a duck” – James Whitcomb Riley
Unless it’s not a duck! This region delves into the world of ciliopathies causing cystic kidney diseases, including mutations in genes other than PKD1 and PKD2 that can cause ADPKD and other ‘polycystic’ kidney diseases such as autosomal recessive polycystic kidney disease, nephronophthisis and nephronophthisis-like disorders, and others!

Copyright: Andrew Glushchenko/ Shutterstock
Introduction
There is growing excitement in the nephrology world about this team. The excitement stems not just from the players, but from one of their coaches, Coach Cilia! Cystic kidney diseases can be classified as inherited vs sporadic, dysplasias vs ciliopathies, or using the Liapis and Winyard classification of kidney cystic disease. Here, we will focus on ciliopathies.
Ciliopathies
Primary cilia are hair-like organelles found on nearly every eukaryotic cell. They are like the coach of the cell. They play a key role in the cellular signaling that governs processes like proliferation, differentiation, and migration during development. Their dysfunction can lead to a broad spectrum of disease affecting multiple organ systems. These diseases are known as “ciliopathies,” and are linked to more than 180 established ciliopathy-associated proteins. In the kidney, ciliopathies are associated with the development of cystic kidney disease.
The most well known kidney ciliopathy is autosomal dominant polycystic kidney disease (ADPKD). ADPKD is the fourth leading cause of kidney failure in the United States. ADPKD has also been an emerging field in terms of treatment. Tolvaptan, a V2 receptor antagonist, is the only current FDA-approved treatment for ADPKD. There are other therapeutic agents currently under investigation, including a microRNA-17 inhibitor, a small molecule corrector for PKD1 variants, sodium-glucose cotransport-2 inhibitors, and metformin, among others.
While ADPKD has been undergoing a renaissance, other rarer causes of ciliopathies have been identified and are similarly poised for therapeutic interventions. These polycystic kidney disease (PKD) masquerader team members include autosomal recessive polycystic kidney disease, nephronophthisis and nephronophthisis-like disorders (Meckel-Gruber syndrome, Joubert syndrome, Bardet-Biedl syndrome), oro-facio-digital syndrome I, tuberous sclerosis, Von Hippel-Lindau Syndrome, as well as medullary sponge kidney. These masqueraders, while occurring less frequently than polycystic kidney disease, are poised to be the next big renaissance in nephrology. See the figure below for some of the known gene mutations that cause these diseases.
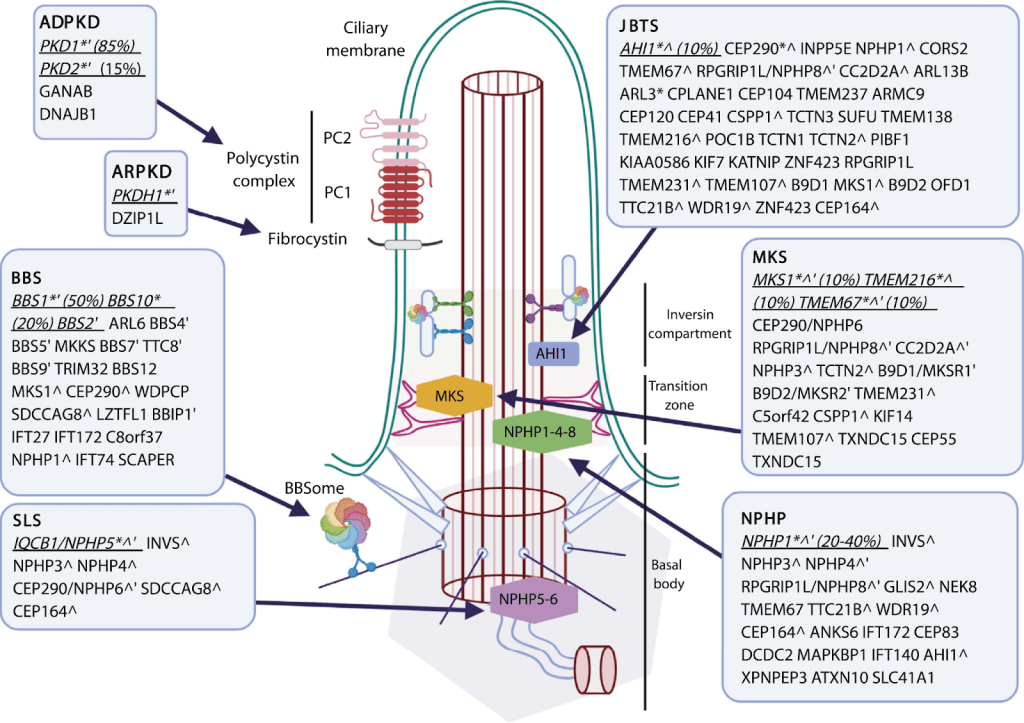
Figure: Renal ciliopathies and associated genotypes. The known reported genes mutated in the renal ciliopathies outlined in this review overlaid on a primary cilium schematic (see Fig 1). ∗Main associated genes. ˆMutated in another renal ciliopathy. ′Part of displayed protein/protein complex. Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease; BBS, Bardet-Biedl syndrome; JBTS, Joubert syndrome; MKS, Meckel-Gruber syndrome; NPHP, nephronophthisis; PC1(2), polycystin 1(2); SLS, Senior-Loken syndrome.
Autosomal Dominant PKD
ADPKD is most commonly caused by mutations to either the PKD1 gene located on chromosome 16 or the PKD2 gene located on chromosome 4. However, recently there have been a slew of additional genes that have been identified. A new “threshold model” of polycystic kidney disease, where cyst formation is triggered by reduced polycystin below a critical threshold in tubular epithelial cells, results in cyst formation. Some of these genes include GANAB, DNAJB11, ALG8, ALG9, PRKCSH, SEC63, and SEC61B, which encode proteins that are important for processing proteins in the endoplasmic reticulum, and trafficking membrane-inserted proteins such as polycystin1 or polycystin2.
ALG8’s story is interesting as it highlights both how far we’ve come in our understanding of genetic diseases and how far we have to go. Biallelic mutations in ALG8 cause a congenital disorder of glycosylation which most commonly presents as hypotonia, protein-losing enteropathy, and liver disease. However, with monoallelic mutations of ALG8, patients can present with both kidney and liver cysts.
Other genes involved include TSC1 and TSC2, which are the primary genes involved in the development of Tuberous Sclerosis Complex. These genes are important regulators of the mTORC1 signaling pathway. Activation of the mTORC1 signaling pathway inhibits polycystin1 and leads to cyst development in Tuberous Sclerosis Complex. TSC2 is located on chromosome 16p13, adjacent to the PKD1 gene, and contiguous deletions to this region results in infantile severe polycystic kidney disease with tuberous sclerosis (PKDTS) contiguous gene deletion syndrome.
In addition, genes such as LRP5, VHL, COL4A1, are also associated with an increased risk of PKD.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Autosomal recessive polycystic kidney disease is typically caused by mutations to PKHD1. This gene encodes multiple, alternatively spliced isoforms to form both membrane-bound and secreted proteins. One of these membrane-bound proteins is the fibrocystin/polyductin complex, which localizes to the primary cilium and centrosome and helps mediate the terminal differentiation of the collecting duct and biliary tract.
ARPKD has an incidence of approximately 1 in 20,000 to 40,000 live births. It is typically identified either in utero or at birth with most fetuses having enlarged, echogenic kidneys and oligohydramnios. The oligohydramnios results in Potter’s sequence, with pulmonary hypoplasia and persistent pulmonary hypertension, which typically drives perinatal mortality in this condition. Of the infants who survive the neonatal period, the mean 5-year survival rate is 85-90%. These patients will develop systemic hypertension, reduced kidney function, hepatic fibrosis, kidney concentrating defects, and portal hypertension. Approximately 29% of individuals will develop ESKD within 10 years, and 58% by 20 years of age. Currently, treatment of ARPKD is supportive, but new therapeutic interventions are on the horizon. Preclinical models overlap in the signaling pathways between ADPKD and ARPKD, which have led to a phase 3 clinical trial for Tolvaptan for ARPKD. There is also growing excitement for the possibility of gene therapy to treat ARPKD.
Nephronophthisis
Nephronophthisis (NPHP) is an autosomal recessive tubulointerstitial kidney disease that typically manifests in childhood. It is one of the most frequent inherited causes of ESKD in children and adolescents. Multiple genes are associated with NPHP, with defects in the gene NPHP1 accounting for ~20% of all cases. Mutations in other NPHP genes account for 1-4% of the remaining NPHP-related disease, meaning that no genetic cause is found in approximately two-thirds of cases. NPHP is also related to other multisystem disorders including Joubert syndrome, Bardet-Biedl syndrome, and Meckel-Gruber syndrome. Children with NPHP will have a slowly progressive decline in GFR with progression to ESKD at a median age of 13 years old. Occasionally, cysts can be detected at the corticomedullary junction or in the medulla. Treatment of NPHP is supportive, but some preclinical trials suggest that treatment with a V2 receptor antagonist slowed kidney disease progression in a mouse model. Other preclinical studies have targeted kidney fibrosis rather than the cystic component of the disease, with low-dose paclitaxel showing promise in rat models.
Medullary Sponge Kidney
Medullary sponge kidney (MSK) is a developmental disorder of the kidneys characterized by dilated medullary and papillary collecting ducts, giving the kidney medulla a spongy appearance. On contrasted CT or IVP, typical findings include a papillary blush pattern that is often described as a bouquet of flowers or paintbrush appearance (see figure below). It typically affects both kidneys but can be unilateral. It is typically a sporadic disorder, but there are some familial clusters that suggest a genetic basis for the disease. MSK is associated with nephrocalcinosis, recurrent nephrolithiasis, renal tubular acidosis, and an increased risk for urinary tract infections. The most common manifestation of MSK is nephrolithiasis, with 70% of patients developing calcium stones. Stone formation is often recurrent and can be characterized by small “sand-like” stone formation. MSK usually presents in the 4th or 5th decade of life. Cysts can be found in the papillae of the affected kidney, and may communicate with the collecting system. The cause of MSK is unknown but there is growing evidence that it may be a ciliopathy, including some genetic studies. Treatment of MSK is supportive, and typically focused on treating hypercalciuria and hypocitraturia for prevention of calcium stones. Treatment with potassium citrate has been shown to decrease the stone event rate and improve bone density in patients with MSK.
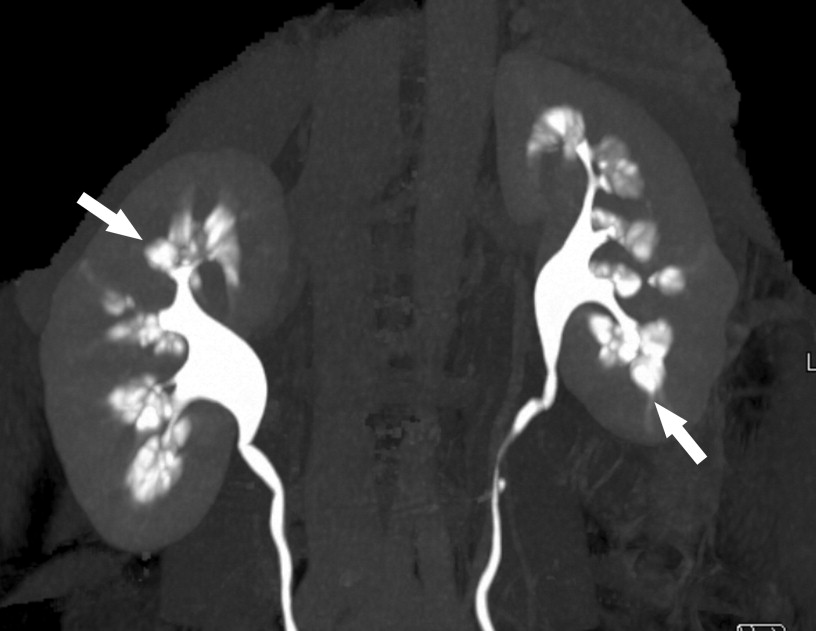
Figure Contrast-enhanced maximum intensity projection 3-dimensional image from multidetector-row computed tomographic urogram shows characteristic papillary blush (arrows) associated with scattered calculi within the dilated collecting tubules. The computed tomographic demonstration is equivalent to the well-documented intravenous pyelography findings.
Autosomal Dominant Tubulointerstitial Diseases
While not classified as a primary ciliopathy, we’d be remiss if we didn’t mention autosomal dominant tubulointerstitial diseases (ADTKD) in this region due to growing links with some of the mutations and cilia. ADTKD is a set of disorders formerly described as medullary cystic kidney disease, UMOD-related kidney disease, or familial glomerulocystic kidney disease with hyperuricemia. ADTKD is typically caused by a mutation in either MUC1, UMOD, REN, HNF1B, or more rarely, SEC61A1. The disease typically manifests in adulthood with slowly progressive tubulointerstitial fibrosis. UMOD, HNF1B, and MUC1 all have ciliary functions, but the primary damage seen is tubulointerstitial fibrosis rather than cilia related. Cysts in patients with MUC1 and UMOD mutations occur at similar rates to all causes of CKD.
HNF1B mutations (or deletions in the case of 17q12 syndrome) again highlight interesting lessons from genetics. Mutations in HNF1B lead to HNF1B-related kidney disease which is also known as renal cysts and diabetes (RCAD) syndrome and HNF1B-MODY or MODY5. HNF1B encodes a transcription factor that regulates gene expression and is widely expressed in multiple fetal tissues with important roles during several stages of nephrogenesis as well as liver, pancreas, and genital tract development. Kidney manifestations are highly variable, even within a family, and include ADTKD, kidney cysts, kidney hypoplasia, single kidney, and other developmental kidney abnormalities such as horseshoe kidney and duplication of collecting system. Hypomagnesemia is also common due to tubular dysfunction. Extra-kidney manifestations include maturity-onset diabetes of the young (MODY), genital tract malformations, abnormal liver function, and gout. In the case of 17q12 syndrome, there can also be neurologic features, including autism spectrum disorders. Treatment is supportive. Sodium-glucose cotransporter 2 inhibitors have been used with success to treat refractory hypomagnesemia in some cases.

Conclusions
A transformative shift in the treatment of polycystic kidney disease and masqueraders is likely coming in the next 5 years. With the growing body of literature connecting genetics to phenotypes, there are many opportunities for targeted therapies and precision medicine to take over from the prior model of supportive care. For conditions like ARPKD, this has already translated into promising clinical trials for Tolvaptan, a therapy initially validated in ADPKD. Similarly, preclinical studies for Nephronophthisis are exploring targeted interventions like V2 receptor antagonists and anti-fibrotic agents, and newly identified ADPKD genes are suggesting targets in protein trafficking. In the next 5 years, improved molecular and genetic knowledge will be crucial for understanding and treating the entire spectrum of polycystic kidney disease.
Team 2: Fabry Treatment
This team represents precision medicine at its finest! Fabry disease is a success story of translational nephrology: genetic diagnosis funnels into actionable, disease-modifying therapy. Here we present the growing pipeline that promises to expand care and diminish limitations of treatments!

Copyright: The Len/ Shutterstock
Fabry’s Starting Lineup: ERT, Chaperones, and Beyond
Why Fabry Matters to the Nephrologist
Fabry disease is an X-linked lysosomal storage disorder caused by mutations in the galactosidase A (GLA) gene. This leads to deficiency of lysosomal enzyme alpha galactosidase A, which normally catalyzes the cleavage of galactose from glycolipids, such as globotriaosylceramide (Gb3). This leads to accumulation of lysosomal Gb3 throughout the body, including the kidneys.
From a nephrology perspective, Fabry disease carries major clinical significance. Kidney involvement typically begins with proteinuria and progresses to CKD and kidney failure in untreated patients. Histologically, characteristic “zebra bodies” – concentric lamellar inclusions seen on electron microscopy (see figure below) – reflect Gb3 deposition in podocytes, peritubular capillary endothelial cells, and other kidney cells. Importantly, by the time kidney manifestations appear, patients almost always have extra-kidney disease, which means that a nephrologist who makes the diagnosis can significantly influence the patient’s overall trajectory. In advanced disease, cardiomyopathy, stroke, neuropathy, and dermatologic complications are common, with cardiac events representing the leading cause of death. Furthermore, patients can experience severe debilitating pain, irritable bowel syndrome-like symptoms, and anhidrosis, which also cause significant morbidity. Finally, there is always the opportunity to screen the patient’s family and initiate treatment early in those affected to prevent loss of kidney function and other manifestations of the disease.
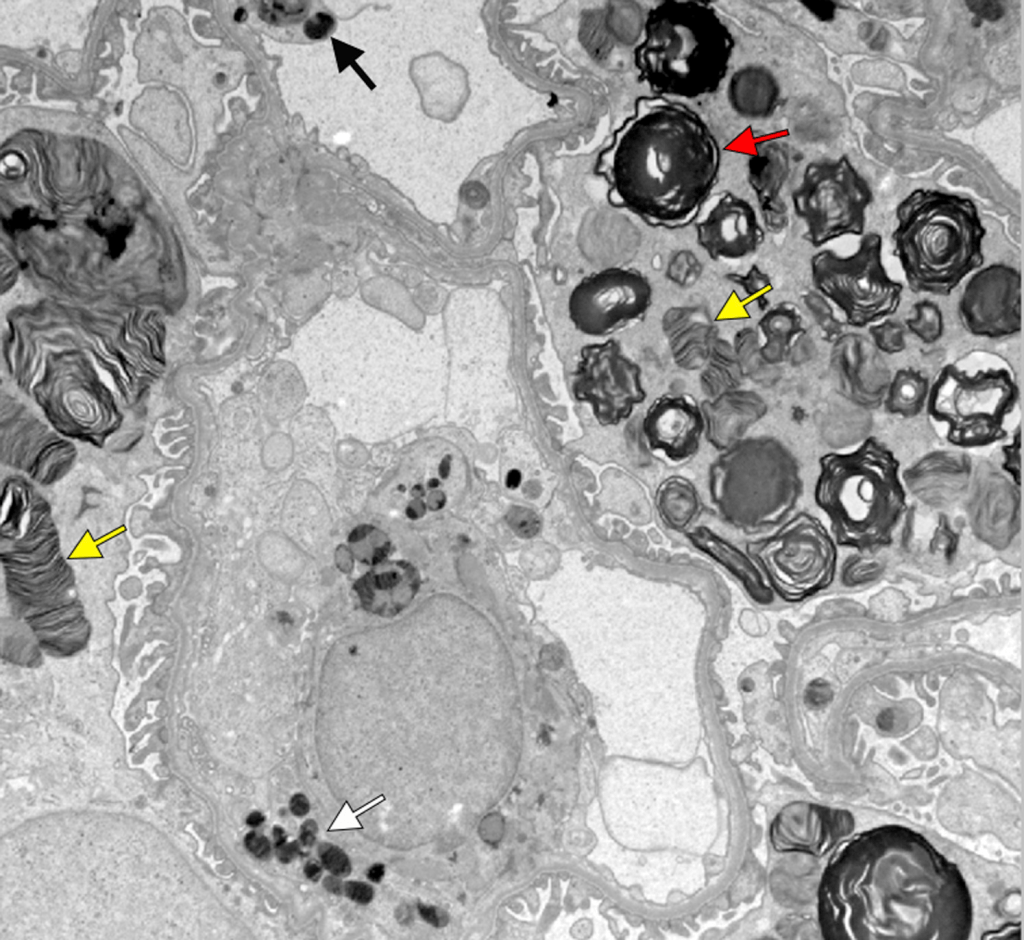
Fabry nephropathy with typical Fabry glycosphingolipid inclusions shaped as multilamellated myelin figures (red arrow) and zebra bodies (yellow arrows) in podocytes. There are also smaller inclusions in endothelial cells (black arrow) and mesangial cells (white arrow; electron microscopy).
The relationship between the genotype and phenotype is central to understanding Fabry disease. Males that are hemizygous for “classic” GLA mutations express less than 1% of mean normal enzyme activity level. These males exhibit Classic Fabry disease and present in childhood typically with abdominal pain and acroparesthesias. Clinical clues include neuropathic pain crises, angiokeratomas, hypohidrosis, cornea verticillata, and unexplained proteinuria or left ventricular hypertrophy in young adults. Diagnosis is typically confirmed by genetic analysis for a pathogenic mutation, low alpha-galactosidase A activity, elevated globotriascylceramide and plasma lyso-Gb3 which serve as a sensitive biomarker of disease burden. By contrast, heterozygous females have unpredictable phenotypes thought to be due to random X chromosome inactivation otherwise known as lyonization. Enzyme activity may fall within the normal range, making genetic testing of the GLA gene essential. While Fabry disease in females is often later-onset and milder, it can be just as severe as the classic male phenotype. Finally, late-onset Fabry disease results from mutations that retain partial enzyme activity. This form typically emerges in adulthood, often with cardiac-predominant manifestations, and lacks the early systemic signs of the classic phenotype – making diagnosis especially challenging. Ultimately, precise genotyping informs treatment, as we’ll discuss below.
The Game Plan: Treatment Options
Advances in therapy have transformed prognosis, making Fabry treatment an exciting team in this year’s tournament.
- Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) has been the backbone of Fabry disease treatment since the early 2000s. The two classic agents produced in mammalian cells – agalsidase alfa and agalsidase beta – are administered intravenously every two weeks, aiming to reduce globotriaosylceramide (Gb₃) substrate accumulation in tissues and thereby slow the progression of organ damage. Early initiation of ERT has been shown to stabilize kidney function, reduce proteinuria, and reduce the size of the left ventricle. However, ERT has its challenges. IV infusions every two weeks impose a high treatment burden and require infusion centers, venous access and associated risks, and infusion-related reactions. Subcutaneous ERT therapy is currently being evaluated. Cost is a major barrier to accessing this therapy. There is also the issue of anti-drug antibody development, which can decrease the effect of ERT should neutralizing antibodies develop. Patients on therapy with acute worsening of kidney function or worsening proteinuria should undergo kidney biopsy to evaluate for accumulation of anti-drug immune complexes in the kidney which can result in a secondary membranous nephropathy and membranoproliferative glomerulonephritis (MPGN). These anti-drug antibodies remain clinically relevant even after kidney transplantation, when ERT is continued to address extra-kidney disease.
Pegunigalsidase alfa is a newer, pegylated ERT produced in plant cell cultures (tobacco leaves!) rather than traditional mammalian cells. Furthermore, this molecule is pegylated with a 2kda peg leading to a longer half life and potentially reduced immunogenicity. In a recent randomized trial, pegunigalsidase alfa showed reduced infusion=related reactions, and comparable results in efficacy when compared to agalsidase beta with respect to the primary endpoint of eGFR decline. However, long-term comparative outcome data (e.g. on ESRD, cardiovascular events) remain limited.

- Chaperone Therapy
Parallel to enzyme replacement, pharmacologic chaperone therapy offers a different mechanism of action. The leading agent in this class is migalastat, an oral small molecule that binds selectively and reversible to certain misfolded forms of alpha-galactosidase A in the endoplasmic reticulum, stabilizes them, and enables their trafficking to lysosomes where the enzyme can degrade its substrate. This approach is only effective in amenable GLA variants (estimated about 35-50% of known mutations). The FACETS trial in treatment-naïve patients and ATTRACT (switch-from-ERT) trial have shown that migalastat achieves reductions in plasma lyso-Gb₃, stabilization of eGFR, and modest reductions in left ventricular mass in eligible patients, with safety comparable to ERT. Because it is oral, migalastat avoids infusion burden and infusion-associated reactions. Nonetheless, its key limitation is that it is not universally applicable, being constrained to the subset of patients whose mutations are responsive; it also requires genetic testing and amenability assay prior to use. Furthermore, it is contraindicated in patients with eGFR of less than 30.

- Substrate Reduction & Gene Therapy: Emerging Draft Picks
Substrate reduction therapy (SRT) and gene therapy represent future draft picks. SRT agents (such as lucerastat and venglustat) aim to reduce the formation of Gb₃ upstream, thereby reducing the burden on whatever residual enzyme activity is present. These are under investigation but not yet approved in Fabry. Gene therapy approaches – ex vivo stem cell harvest and modification versus in vivo adenovirus-mediated gene transfer – offer the possibility of durable, perhaps one-time, treatment. Early preclinical work and limited early human trials suggest that sustained alpha-galactosidase expression may be possible, but important questions remain regarding safety (e.g., vector immunogenicity, insertional risk), durability, and control of expression levels.
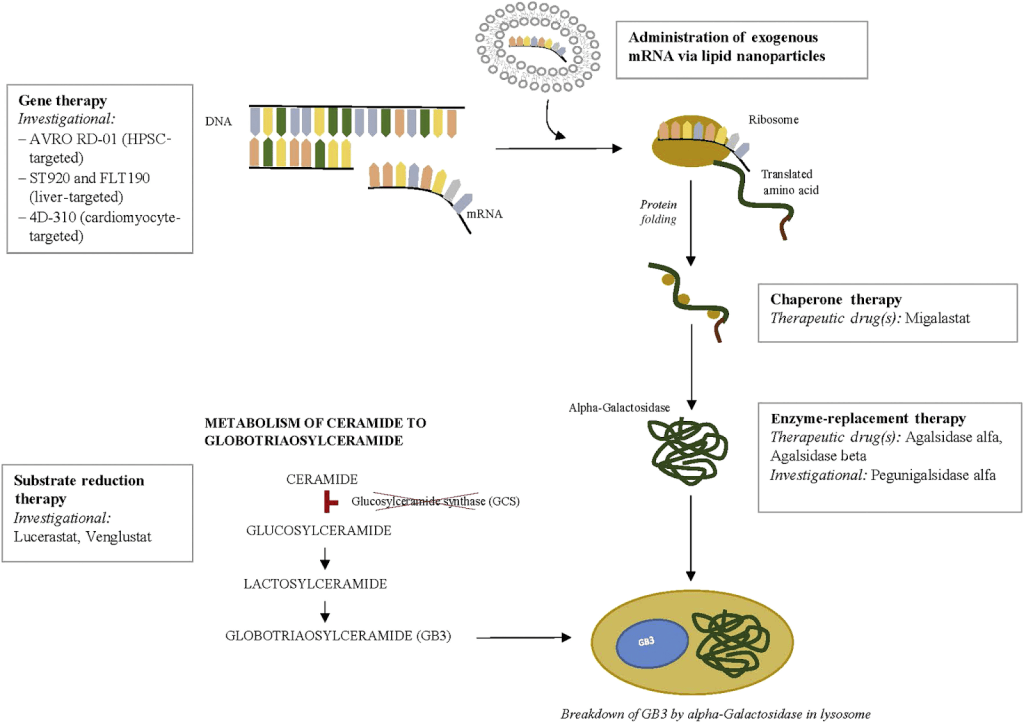
Figure: Current and investigational therapeutic agents for Fabry disease are depicted at each potential point of therapeutic intervention. As shown, therapies are directed at either replacing or generating deficient enzyme, or blocking the accumulation of substrate. Clinical trials with gene therapy approaches to the treatment of Fabry disease are ongoing. Chaperone therapy is now available for amenable mutation with migalastat. Enzyme replacement therapy remains the mainstay of treatment for most patients with Fabry disease, with agalsidase α and agalsidase β, while pegunigalsidase is in clinical trials. Substrate reduction therapy (SRT) consists of glucosylceramide synthase inhibitors and aims to reduce the accumulation of toxic substrate; SRT agents are currently in clinical trials.
- Adjunctive Therapies
Treatment of Fabry disease does not just involve treatment around the deficiency of alpha-galactosidase A or the accumulation of lysosomal Gb3. General CKD care, treatment of symptoms, and replacement of nutritional deficiencies are all important. Treatment with medications to reduce proteinuria such as sodium-glucose cotransporter-2 inhibitors and inhibitors of the renin-angiotensin system are both important and effective. Neuropathic pain can be treated with carbamazepine and neurontin. Vitamin D deficiency is common in patients with Fabry disease and should be repleted. Those with overt vitamin D deficiency have a higher risk of cardiomyopathy and worse proteinuria. It is not yet known if repleting vitamin D reverses these findings.
Strengths on the Court
Together, this multi-modality treatment approach represents both strength and complexity in the Fabry disease playbook. The strengths of this strategy include both the possibility of improved survival and quality of life, particularly when therapy is started early, before irreversible organ damage ensues. Newer options such as chaperone therapy and gene therapeutics are vivid examples of how basic science is informing genotype-driven decision making in rare diseases.
Weaknesses and Fouls
A major obstacle is late diagnosis; many patients only receive a diagnosis once proteinuria or CKD is underway, limiting reversibility. Treatment burden and cost are non-trivial: lifelong infusions (or chronic therapy) in the face of expensive drugs place strain on patients and payors, and access is inequitable globally. Efficacy is also variable – not all disease manifestations reverse under treatment. As we think about Fabry in the transplant population, ERT neutralizing antibodies loom large. In gene therapy, unknowns are still substantial. In clinical research, the ideal endpoints for monitoring success (e.g., eGFR slope, lyso- Gb₃, biopsy substrate burden, hard clinical outcomes) remain the subject of debate.
Why this Team Could Go All the Way
Fabry disease is already a success story of translational nephrology: genetic diagnosis (the first team) funnels into actionable, disease-modifying therapy (the second team). The growing pipeline promises that many of the current limitations may soften over time. And symbolically, Fabry disease treatment underscores nephrology’s role in rare disease care, precision medicine, and the translation of molecular insight into therapeutic impact.
Join hosts Matthew Sparks and Samira Farouk as they chat with NephMadness writers Steven Clapp & Alyssa Steitz, and Director of Rare Genetic Kidney Diseases at the University of Alabama Birmingham, Eric Wallace. How can we spot the PKD wanna-bes? What’s new in the treatment of Fabry disease? And how do you say Fabry? Fab-ray? Fab-ree?
Episode 17: NephMadness 2026 & Genetics & PKD Masqueraders & Fabry

– Executive Team Members for this region: Anna Burgner @anna_burgner – @annaburgner.bsky.social and Samira Farouk @ssfarouk.bsky.social | Meet the Gamemakers
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC per region through NKF PERC (detailed instructions here). The CME and MOC activity will expire on June 1, 2026.
Leave a Reply