
#NephMadness 2026: Trolls of Transplantation Region
Submit your picks! | @NephMadness | @nephmadness.bsky.social | NephMadness 2026
Selection Committee Member: Sam Kant @kantsmd
Sam Kant is an Nephrologist & Transplant Physician at St. Vincent’s University Hospital and University College Dublin, where he is also the Director of the Internal Medicine Training Program in conjunction with the Royal College of Physicians of Ireland.
Selection Committee Member: Jeannina Smith @germatologist.bsky.social
Dr. Jeannina Smith is a Professor (CHS) at the University of Wisconsin School of Medicine and Public Health. She is the Medical Director of the Transplant Infectious Disease Program. Her research focuses on infections in solid-organ transplant recipients, with a focus on viral infections including COVID and CMV, fungal infections, and antifungal pharmacology. She is passionate about medical education and is the former program director of the infectious disease fellowship and currently directs a preclinical integrated block at UW School of Medicine and Public Health and received the 2021 Dean’s Teaching Award for her teaching contributions.
Writer: Hariharasudan Natarajan @HariNataraj94 – @beanbuddy.bsky.social
Hariharasudan Natarajan is a second-year nephrology fellow at Mount Sinai Hospital. He completed his internal medicine residency at the University of Oklahoma. His clinical interests lie in transplant nephrology, with a particular focus on recurrent glomerulonephritis affecting kidney allografts.
Writer: Caitlyn Vlasschaert @DrFlashHeart – @drflashheart.bsky.social
Caitlyn Vlasschaert is a nephrology fellow at Queen’s University in Canada. She completed a PhD during internal medicine residency studying how clonal hematopoiesis affects the kidneys and is broadly interested in nephrogenetics.




Competitors for the Trolls of Transplantation Region
“I’ll be back.” — Arnold Schwarzenegger (The Terminator, 1984)
BK virus (BKV) and cytomegalovirus (CMV) are everywhere. The majority of us have been exposed to these trolls and carry them quietly as they lie hidden in our cells. Immunosuppression in a kidney transplant recipient can awaken these beasts and allow them to wreak havoc. This region dives into these trolls’ caves where these viruses live, tries to uncover their secrets, and outlines potential future strategies to slay them once and for all.
Team 1: BK
versus
Team 2: CMV
Image generated by Matthew Sparks using ChatGPT at http://chat.openai.com, February 2026. After using the tool to generate the image, Sparks and the NephMadness Executive Team reviewed and take full responsibility for the final graphic image.
Coming in (Quietly) Off the Bench
BK virus (BKV) and cytomegalovirus (CMV) spend most of their lives sitting quietly on the bench, barely noticed. Immunosuppressive therapy used in transplantation unfortunately gives them their chance to check in – and when they do, they can dominate the entire game. These viruses don’t just test antiviral strategies; they can challenge how we balance immune system control against graft survival. Here, we size up both challengers, review their biology and playbooks, and examine how clinicians fight back.
Team 1: BK
“You can’t kill the boogeyman.” — Laurie Strode (Halloween, 1978)
BK virus, named for the Sudanese patient from whom it was first isolated, lurks in the shadows of urothelial and tubular cells. You won’t even find it unless you specifically go looking. Even though it can cause kidney transplant failure, Team BK won’t even cause any symptoms – and when you do find it, options are close to nothing (…for now) if lowering immunosuppression doesn’t do the trick.

Copyright: Sakis Lazarides/ Shutterstock
BK Virus: A Silent Passenger With a Heavy Toll
Look out for the ubiquitous BKV on the court, a polyoma virus that nearly all of us carry quietly as a silent, dark passenger. BKV was first identified in 1971 in a Sudanese kidney transplant recipient (B.K.) in London with ureteric obstruction and graft dysfunction. Like its cousin JC virus (similarly named for John Cunningham, from whom it was first isolated), BKV had likely co-existed with humans for millennia, but only became clinically visible when transplant medicine created the right immunosuppressive conditions. By adulthood, up to 90 percent of people worldwide are seropositive. Primary infection usually occurs in childhood, spreads most commonly via the respiratory tract, and causes a short viremia. During this viremic phase, the virus seeds the kidney, where uroepithelial and tubular cells express α2,8-linked sialic acids and gangliosides that BKV uses as its entry receptors. Once inside, it establishes lifelong latency in an episomal form – i.e., as extra-chromosomal circles of DNA tucked into the nucleus without producing viral proteins, a literal sleeper. In this essentially hidden state, CD8⁺ T-cells cannot recognize or clear it. For most individuals this latency is harmless, but under immunosuppression after kidney transplantation, BKV can awaken like a defender who comes out of nowhere. The consequences can be serious: tubulointerstitial nephritis, ureteral strictures, and most concerningly, allograft loss. BKV nephropathy occurs in ~1-10% of kidney transplant recipients and results in graft loss in 15-50% of cases, particularly when diagnosis is delayed.
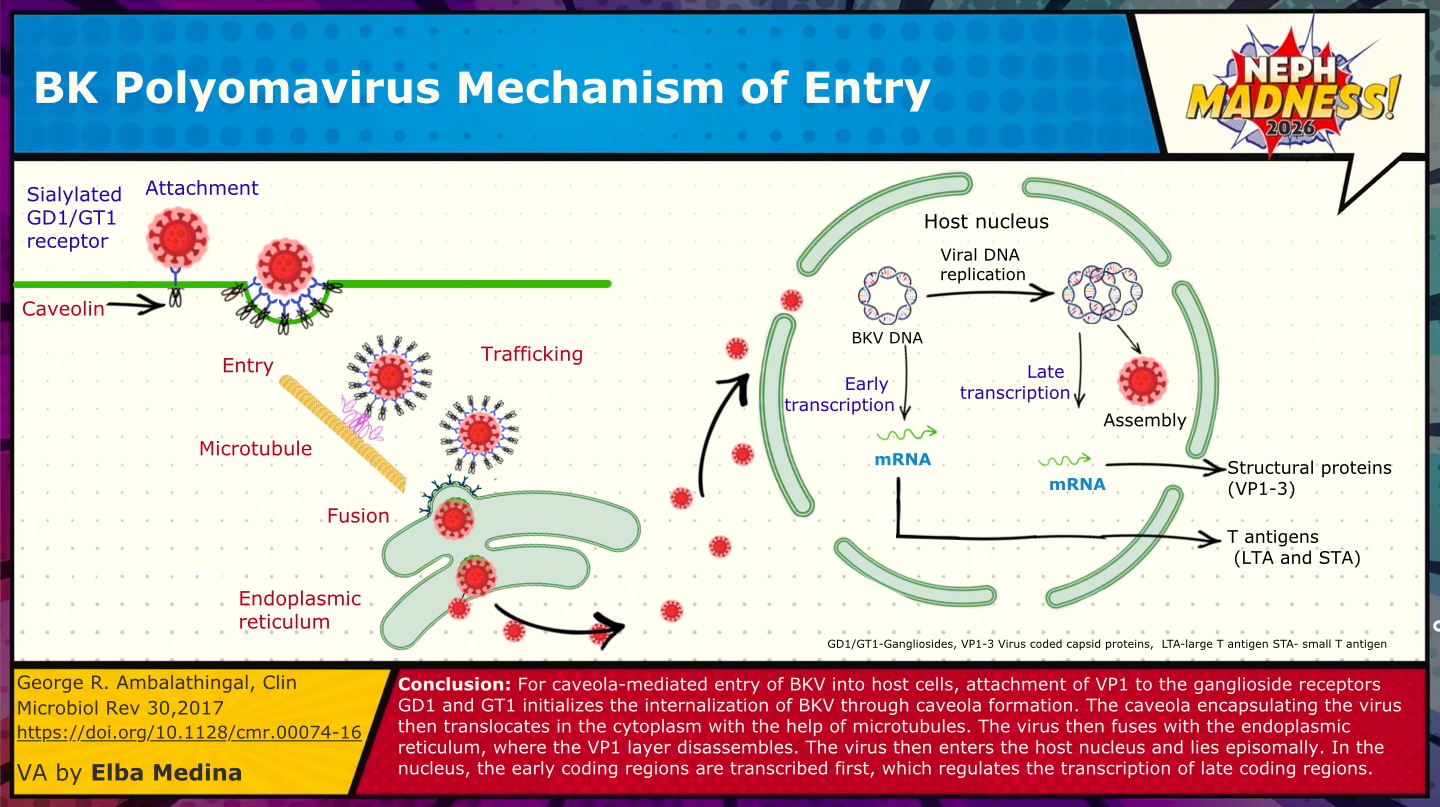
Who’s At Higher Risk?
Risk factors for BKV reactivation and associated nephropathy reflect a multifactorial interplay between donor, recipient, and transplant characteristics, though, not surprisingly, the overall intensity of immunosuppression remains the single strongest determinant. Emerging biomarkers such as torque teno virus (TTV) – a harmless, commensal virus whose circulating levels reflect the overall strength of the immune system – may help refine risk assessment by providing a quantitative measure of each patient’s net immunosuppressive load.
On the recipient side, older age, male sex, prior kidney transplantation, and recent rejection episodes have been consistently linked to BKV reactivation. Host immunogenetics also play a role: specific human leukocyte antigen (HLA) class I and II profiles, along with variants in interferon-γ and HLA-E genes, may influence the strength and breadth of antiviral immune responses, subtly shaping individual susceptibility.
When to Look for BKV
Not surprisingly, the earliest signals of reactivation are BKV viruria and viremia, which often precede nephropathy by months (Note: what we measure in clinical practice is NOT viremia or copies of the actual intact virus, but the viral DNA – to be more precise “DNAemia” or “DNAuria”). The Second International Consensus Guidelines on the Management of BK Polyomavirus in Kidney Transplantation suggest plasma DNA screening for the first two years of transplantation. When testing is unavailable, urine cytology can be used to look for “decoy cells” (uroepithelial or tubular cells containing BK viral inclusion bodies). That being said, BKV can reactivate at any time during one’s kidney transplant journey – so look for it whenever there is evidence of allograft dysfunction: it’s the “B” in CRAB, a mnemonic for the differential diagnosis for acute kidney injury of the allograft. The recent guidelines remind us that early detection is key and provide a critical window to safely adjust immunosuppression, and thereby prevent progression to BK virus nephropathy and potential graft loss.
What to Do When BKV Rears Its Ugly Head
When low levels of BKV viremia are detected, the first step is to keep calm and confirm persistence with repeat testing in 1–2 weeks. If viremia exceeds more than 10,000 copies, a presumptive diagnosis of BKV-associated nephropathy (BKVAN) is made. Viral loads over this threshold have a nearly 90% specificity for BKVAN. As one would do with any other infection, step one is immunosuppression reduction. A kidney biopsy is typically not performed to confirm the diagnosis, though is recommended in specific situations:
- Persistent or high-level viremia with rising creatinine or other evidence of graft dysfunction;
- Children and high-immunologic-risk recipients, even if renal function appears stable;
- To exclude concurrent rejection, particularly if immunosuppression has already been reduced.
When a kidney biopsy is performed, the diagnosis of BKVAN is established by demonstrating viral cytopathic changes in tubular epithelial cells, typically characterized by enlarged nuclei with basophilic intranuclear inclusions. Confirmation requires immunohistochemical staining for the SV40 large T antigen, whose amino acid is 75% identical to the BKV large T antigen (also similar to the JCV T antigen) and thus has cross-reactivity. Histologically, BKVAN manifests as tubulointerstitial nephritis with varying degrees of tubular injury, interstitial inflammation, and fibrosis, often in a patchy distribution. Viral cytopathic changes may not always be visible on light microscopy, and distinction between other causes of tubulointerstitial nephritis, like acute cellular rejection, and BKVAN can be a challenge. In these cases, viral immunohistochemistry (IHC) stains like SV40 are critical to decide if immunosuppression should be increased or decreased. For pathology enthusiasts, the BKVN histologic findings can be categorized into three classes, with higher classes being associated with poorer outcomes.

A) (Left) Dark purple (basophils) inclusions in tubular epithelial cells suggest BKV associated nephropathy. B) (Right) Immunohistochemistry stain for SV40 confirms the presence of BKV within these inclusions. Adapted from the AJKD Atlas of Renal Pathology article on Polyomavirus Nephropathy.
Again, the cornerstone of management remains judicious reduction of immunosuppression, guided by viral load, graft function, and histologic findings. The goal is to restore antiviral immunity without provoking rejection; a careful recalibration rather than a withdrawal.
The 2024 International Consensus Guidelines on the management of BKV in kidney transplant (over 20 pages) outline two acceptable reduction strategies: one beginning with antimetabolite reduction by 50%, the other with calcineurin inhibitor target level reduction. In both, the goal is a 10-fold drop in plasma BK viral load within 4 weeks or full clearance below the assay’s lower limit of detection.
After confirmed clearance of BK viremia, maintenance immunosuppression should be cautiously re-escalated according to the patient’s individual immunologic risk (e.g., presence of donor- specific antibodies [DSA], prior episodes of rejection), with continued periodic screening for recurrent viremia.
Reduction of immunosuppression is effective in most cases: viremia resolves in over 80% of patients if detected early, though less often among those who develop BKVAN. However, acute cellular or antibody mediated rejection can occur in up to 15% – which should serve as a reminder to lower immunosuppression thoughtfully. The timing of intervention matters greatly: when BKV is recognized during asymptomatic viremia, most grafts can be preserved. Once nephropathy is established, however, long-term graft survival declines substantially, with 30–50 percent allograft loss within 3–5 years despite intervention.
Why Antivirals Don’t Work
So why do we treat BK virus by simply reducing immunosuppression rather than using an antiviral drug? How about an effective offensive strategy? Over the past two decades, a variety of pharmacologic approaches have been tested, but unfortunately none have shown consistent benefits in randomized or well-controlled studies. These agents were pursued because of early in vitro or observational signals.
| Cidofovir | A nucleotide analogue originally developed for cytomegalovirus, cidofovir was found to inhibit BKV replication in cell culture, leading to its off-label use in kidney transplant recipients. A 2010 meta-analysis found that cidofovir offers no improvement in graft survival and carries a significant risk of nephrotoxicity |
| Leflunomide | An immunomodulatory and antiviral pyrimidine-synthesis inhibitor, leflunomide appeared to have anti-BKV activity in cell culture and some supportive retrospective data. However, many studies report contradictory results, and leflunomide has not clearly demonstrated superiority over standard immunosuppression reduction alone. |
| Fluoroquinolones | Fluoroquinolone antibiotics were investigated after laboratory evidence suggested they interfere with the BKV large T-antigen helicase. Randomized controlled trials of levofloxacin or ciprofloxacin prophylaxis, however, did not reduce BK viremia and instead increased antimicrobial resistance. |

These failures reflect the unique biology of BKV: a non-enveloped, circular, double-stranded DNA virus with an exceptionally stable icosahedral capsid and NO virus-specific polymerase or protease (the usual targets for antiviral drugs). Once established, it persists episomally within long-lived uroepithelial and tubular cells, reactivating only when immune surveillance falters. Thus, effective control depends not on drug inhibition of viral enzymes, but on the reconstitution of T-cell–mediated immunity. Neutralizing antibodies, though present in nearly all adults, do not prevent reactivation because serologic immunity alone cannot access the intracellular, latent form of the virus.
Therefore, the 2024 Guidelines explicitly recommend against the use of cidofovir, leflunomide, or fluoroquinolones, for either prevention or treatment of BK viremia or BK virus nephropathy. Intravenous immunoglobulin (IVIG) may be considered as adjunctive therapy in patients who fail to clear viremia or in those at high immunologic risk who require substantial immunosuppression reduction and can be effective; however, there is a lack of randomized controlled data (sensing a theme?). A recent systematic review found that while IVIG infusions may improve BK viremia, their impact on allograft function and survival is “uncertain.” Consequently, careful, stepwise reduction of immunosuppression remains the only consistently effective and evidence-based treatment strategy available today. Though sometimes a challenge for those practicing medicine, BKVAN might be a prime example of when to practice masterly inactivity (pending potential developments below).
Potential Future Diagnostic & Therapeutic Options
Despite major advances in detection and management, BKV remains a biologically elusive and clinically frustrating adversary. Biopsy with SV40 immunohistochemistry continues to define the diagnostic gold standard, yet its invasiveness and patchy sensitivity underscore the need for better noninvasive tools. Donor-derived cell-free DNA (dd-cfDNA) assays now show promise as adjuncts capable of distinguishing viral injury from alloimmune rejection.
At the same time, advances in humoral and cellular immunotherapy are reshaping how we think about BKV control. The virus exists as four major genotypes that are now known to represent four distinct serotypes, and low pre-transplant neutralizing antibody titers against the donor’s serotype have been linked to higher post-transplant viremia. These insights have renewed interest in immune-based strategies that selectively enhance BKV-specific immunity without broadly compromising graft function. For example, virus-like particle (VLP) vaccines against BKV have demonstrated robust neutralizing antibody responses in preclinical models and represent a compelling preventive strategy, particularly in the pre-transplant setting.
Parallel efforts in passive immunotherapy include monoclonal antibodies targeting the VP1 capsid protein, the sole viral surface protein mediating host-cell attachment. Early-phase trials of high-affinity neutralizing antibodies such as potravitug and MAU868 have demonstrated favorable safety profiles and sustained receptor occupancy, supporting their potential use as preemptive or adjunctive therapies for BK viremia. In parallel, virus-specific T-cell (VST) therapies attempt to replace the BK-targeted immunity lost under immunosuppression by infusing donor-derived T-cells trained to recognize BK virus. For example, posoleucel is an allogeneic VST which targets not only BKV, but adenovirus, CMV, Epstein_Barr virus, human herpesvirus 7, and JCV – six for the price of one. Posoleucel comes from the peripheral blood mononuclear cells of seropositive healthy donors, whose cells are then expanded after culture with viral peptides. A 2024 phase two study found that it was safe and associated with a larger reduction of BK viremia compared with placebo. The goal is to restore antiviral control without broadly increasing immunosuppression, though questions remain regarding safety, cost, and how often cells need to be given.

Beyond immune-based approaches, emerging therapies target essential components of BK virus replication, including agnoprotein (needed for critically important viral functions: assembly, maturation, and viral release) and viral mRNA splicing. There is more work to be done. Thirteen trials are actively recruiting patients with BKV, and include mechanistically promising studies of BKV-specific cytotoxic T-cells. Here’s to hoping that one of these treatments will eventually make it to the big stage to once and for all slay this pesky troll that loves to hideout – and until then, we’ll continue on with the KISS approach: keep it simple, stupid (and just lower the immunosuppression). But Team BKV is scrappy – just check out this Reddit thread titled “BKV Won’t Go Away.” Watch out for this yet-to-be-tamed opponent to steal this year’s NephMadness crown.
Team 2:
CMV
“Who’s that tripping over my bridge?” — The Troll (The Three Billy Goats Gruff, 1840s)
Cytomegalovirus (CMV, AKA human herpes virus 5), commonly known for the “owls’ eye” inclusion that can be seen on histology, is the most common opportunistic infection seen in kidney transplant recipients. Some may even call it the GOAT of herpes viruses given its propensity for genomic alterations which can make treatment of resistant CMV a real doozy. Prevention is key, but can also come at a cost. Sometimes CMV makes its presence clearly known, but like any good troll, it also knows how to be a menace in the dark.

Copyright: Wirestock Creators/ Shutterstock
CMV: Whether Asymptomic or Deadly, Always Harmful
Discovered in 1956 in the salivary glands of two terminally ill infants, CMV has been on the solid organ transplantation scene since 1965 when it was first identified in a kidney transplant recipient. Since then, it has asserted itself as a tough opponent on the court as the most common opportunistic infection among kidney transplant recipients. CMV infection has been associated with increased mortality and morbidity, as well as accelerated graft failure post-transplant. Newer therapies including small molecules, T-cell therapies, vaccines, and monoclonal neutralizing antibodies are slowly emerging and provide some hope to hopefully definitively overcome this traditionally powerful “troll” and improve kidney transplant outcomes.
Also Known As Human Herpes Virus 5
CMV, less commonly referred to as Human Herpes Virus 5, is a beta herpes virus belonging to the family Herpesviridae. Its virion consists of an inner genome core, capsid, and envelope. Glycoproteins on the envelope, potential targets for neutralizing antibodies, are used for cellular entry after binding to receptors on human cells. The CMV genome (which is transported to the cell nucleus) encodes its own DNA polymerase, another drug target. Like all human herpesviruses, CMV is a linear double-stranded DNA virus. Its genome might be the GOAT of herpes viruses: the largest, with the highest level of genetic diversity of all the known human herpesviruses due to frequent genomic recombination and “superinfections” with other CMV viruses. This propensity for genomic alteration has important implications for treatment, particularly its resistance to antiviral agents. Think of CMV like Michael Jordan – constantly adapting its moves to evade the defense, and then becoming impossible to defend against. CMV produces a number of proteins which protect the hijacked cells from apoptosis and allow evasion of immune medicated destruction, including modulation of antigen presentation and processing, alteration of cytokines, and evasion of innate immunity.
Viral gene products are also used to make new infectious viruses which are packaged in envelopes and leave the cell (facilitated by pUL97, not 6-7) to infect nearby uninfected cells. After primary infection, the virus is thought to reside in cellular reservoirs including hematopoietic stem cells, though these reservoirs are somewhat controversial. Herpes viruses are very common infections infecting the majority of humans by adulthood and they are often acquired in childhood and acquired from other infected humans (with no animal reservoir). Infectious persons often have no symptoms, and they establish lifelong latent infections which bring to life the old expression, “Love may not last forever. But herpes does.”
The life cycle of human herpes viruses is divided into two phases: the lytic phase where there is active viral replication and contagion, and the latent phase or viral “hibernation.” Like herpes simplex virus (HSV) 1 and 2, CMV can be woken from this slumber. Viral reactivation can cause recurrent infection and infectivity with or without symptoms.
A Ubiquitous Pathogen
Primary infection with CMV often occurs early in life, and it is estimated one in three American children have CMV by the age of five and over half of US adults have been infected with CMV by age 40. Seroprevalence increases with age and is notably more common in deceased organ donors than in potential recipients. Transmission can occur through close contact with human bodily fluids such as saliva, urine, genital secretions, as well as vertical transmission, transfusion, or transplantation. Infection and reactivation elicit strong humoral and cell-mediated immune responses. This response protects most immunocompetent patients from developing recurrent symptoms of infection in the setting of reactivation and end organ disease is extremely rare. However, the immunosuppressive therapy used post-transplant to prevent rejection also blunts the immune responses to CMV, especially cell-mediated immunity, and can lead to active CMV replication and tissue damage. With some preformed immunity this destructive effect is blunted, but if CMV is acquired for the first time while the individual is immunosuppressed, the typical development of this protective immune response is blunted and in this setting the individual is at highest risk of CMV end organ disease. CMV is also noted to be immunomodulatory: CMV infection in transplant recipients is associated with a heightened risk of opportunistic infection, particularly pneumocystis and Aspergillus infection. Interestingly, CMV infection can also induce inflammation within the allograft, leading to rejection and poor graft survival through several immunomodulatory mechanisms. In summary, CMV infection after transplant places patients at risk of infection and rejection, two foes which typically play for opposing teams!
How To Find CMV
CMV infection is defined as the presence of CMV replication in the body, and this can be asymptomatic. CMV replication can be detected by multiple methods including pathology assessing typical cytopathic effect, viral culture, or antigenemia assay but in the modern era is most typically assessed seeking CMV nucleic acid by a CMV-specific polymerase chain reaction (PCR) assay which has been standardized. If CMV replication is associated with systemic signs and symptoms—such as fever, neutropenia or thrombocytopenia, and malaise (CMV syndrome)—or tissue-invasive disease, it is characterized as CMV disease. The PCR-based assay has a higher sensitivity and similar specificity to the antigenemia assay. While CMV tissue invasive disease is almost always accompanied by CMV viremia, typically high viral load levels, tissue-invasive diseases (including gastrointestinal disease, pneumonia, hepatitis, pancreatitis, encephalitis, nephritis, cystitis, mucocutaneous disease, myocarditis, and chorioretinitis) are not always associated with positive blood PCR. In those cases, strong clinical suspicion, along with pathology or cytology testing, may be required. CMV serology is not helpful for disease diagnosis, as primary CMV infection and hence seropositivity (IgG) is highly prevalent; however, it is used in pretransplant evaluation to assess the risk of post-transplant CMV reactivation and disease based on the serostatus of donor (D) and recipient (R). Risk levels are stratified into three categories: high (D+/R-), intermediate (D+/R+ or D-/R+), and low (D-/R-) for the reasons noted above.
And a few words about CMV-related kidney disease (this is NephMadness). Findings mimic Team BKV: tubulointerstitial nephritis, with classic intranuclear glassy, basophilic inclusions with a surrounding halo (“owl’s eye”). A CMV IHC stain can confirm the diagnosis. CMV-associated collapsing focal, segmental glomerulosclerosis has also been reported.
An Ounce of Prevention is Worth a Pound of Cure?
Why is a preventative strategy important? In the absence of a preventative game plan, 40% to 100% of all kidney transplant recipients develop CMV infection, and up to 67% develop CMV disease. Current protocols reduce the risk to 17% to 92% and 0% to 37%, respectively. Individuals classified as low risk (Donor CMV IgG – /Recipient CMV IgG -) have an incidence of <5% for CMV disease and can be monitored clinically. There are two preventative approaches for individuals at risk: universal prophylaxis and preemptive therapy.
Universal Prophylaxis
For universal prophylaxis, all patients are initiated on anti-viral drug for a finite duration post-transplant. For many years, valganciclovir was the MVP for prophylaxis due to its ease of oral dosing, potency and clinical efficacy. It is typically continued for 3 months in the intermediate-risk group and 6 months in the higher-risk group (D+/R-). The weakness of this player is its toxicities, particularly to the bone marrow; leukopenia often necessitates discontinuation of other important medications such as sulfa prophylaxis or mycophenolate. The fact is that very potent prophylaxis can delay but not prevent CMV disease, and in the absence of the individual’s development of CMV-specific immunity, pharmacologic prophylaxis has been described as the “kick the can down the road” approach. Because of these limitations, a new hot shot has recently joined this roster: letermovir, a viral terminase inhibitor, has demonstrated and was recently approved by the FDA for prophylaxis. This treatment is taking the league by storm. It can reverse the leukopenia seen with valganciclovir and prevent the reduction of mycophenolate associated with rejection. A 2023 study found that letermovir was non inferior to valganciclovir for prophylaxis of CMV disease, with lower rates of leukopenia. Additionally, it may allow more robust development of CMV specific cellular immune response. Letermovir does have an important handicap: it does not treat herpes and the addition of acyclovir or valacyclovir is necessary.

Preemptive Therapy
The second approach is preemptive therapy. This game plan is predicated by the understanding that low level viremia typically progresses to higher levels of viremia before symptoms, and — most critically — tissue invasive disease. To that end, preemptive monitoring seeks to find viremia at low levels and treat it before it can do damage. This involves serial CMV DNAemia testing (usually performed weekly) to detect asymptomatic viral replication and initiate treatment at a prespecified threshold before it has a chance to cause disease. In large trials, no difference in the incidence of CMV disease or graft loss was identified in comparison to universal prophylaxis. Although it comes with the benefit of a lower pill burden and drug toxicities, it comes with logistical challenges such as weekly testing, timely review of results, and initiation of treatment. This method also allows the individual to develop CMV-specific immunity which is helpful in the longer run. Currently, there is no role for CMV immunoglobulins in prophylaxis, especially given the availability of potent antivirals.
The duration of prophylaxis can also be personalized using assays that measure CMV-specific T-cell-mediated immunity, such as CMV-specific interferon-gamma enzyme-linked immunosorbent spot (ELISpot) or Quantiferon CMV assay, every 4 weeks from day 30 after transplant. This approach has shown a clinically significant reduction in the duration of antiviral prophylaxis and, consequently, neutropenia without increasing the risk of CMV disease. However, results are less robust for outcomes including CMV replication.
Some Treatments That Work and Those That Don’t
The treatment of CMV should always include two elements: reduction of immune suppression and antiviral therapy. Our understanding of CMV and potential targets has led to the development of antivirals. The exact drug selected and route of administration depends on the immunological risk of the individual, severity of the disease and the viral load. First-line options include oral valganciclovir (900 mg twice daily, adjusted for renal function) and intravenous (IV) ganciclovir (5 mg/kg daily, adjusted for renal function), both of which are activated by the viral kinase pUL97 and work by inhibiting DNA polymerase pUL54. The VICTOR trial demonstrated the non-inferiority of oral valacyclovir to IV ganciclovir in the management of non-life-threatening CMV disease, making it the preferred treatment option for this patient population. In the case of life-threatening or sight-threatening disease or when a high viral load is present, the consensus is to initiate treatment with IV ganciclovir to ensure optimal drug exposure. Whenever possible immunosuppression should also be lowered. Treatment can be transitioned to oral Valacyclovir after clinical improvement, if oral therapy can be tolerated. Viral load monitoring should be performed weekly, and treatment should be continued until the DNAemia has decreased below the laboratory or institutional threshold on two consecutive weekly samples. Despite the lack of evidence supporting the use of secondary prophylaxis in reducing the risk of relapses, experts have used oral Valacyclovir for 1-3 months in patients deemed at high risk of relapse. Switching to mTOR inhibitors has been shown to reduce the recurrence of CMV: mTOR inhibitors may be considered in conjunction with belatacept for high-risk CMV seropositive recipients in whom a transition to belatacept is planned, as belatacept alone has been reported to deplete CMV-specific cell-mediated immunity and induce late CMV disease.
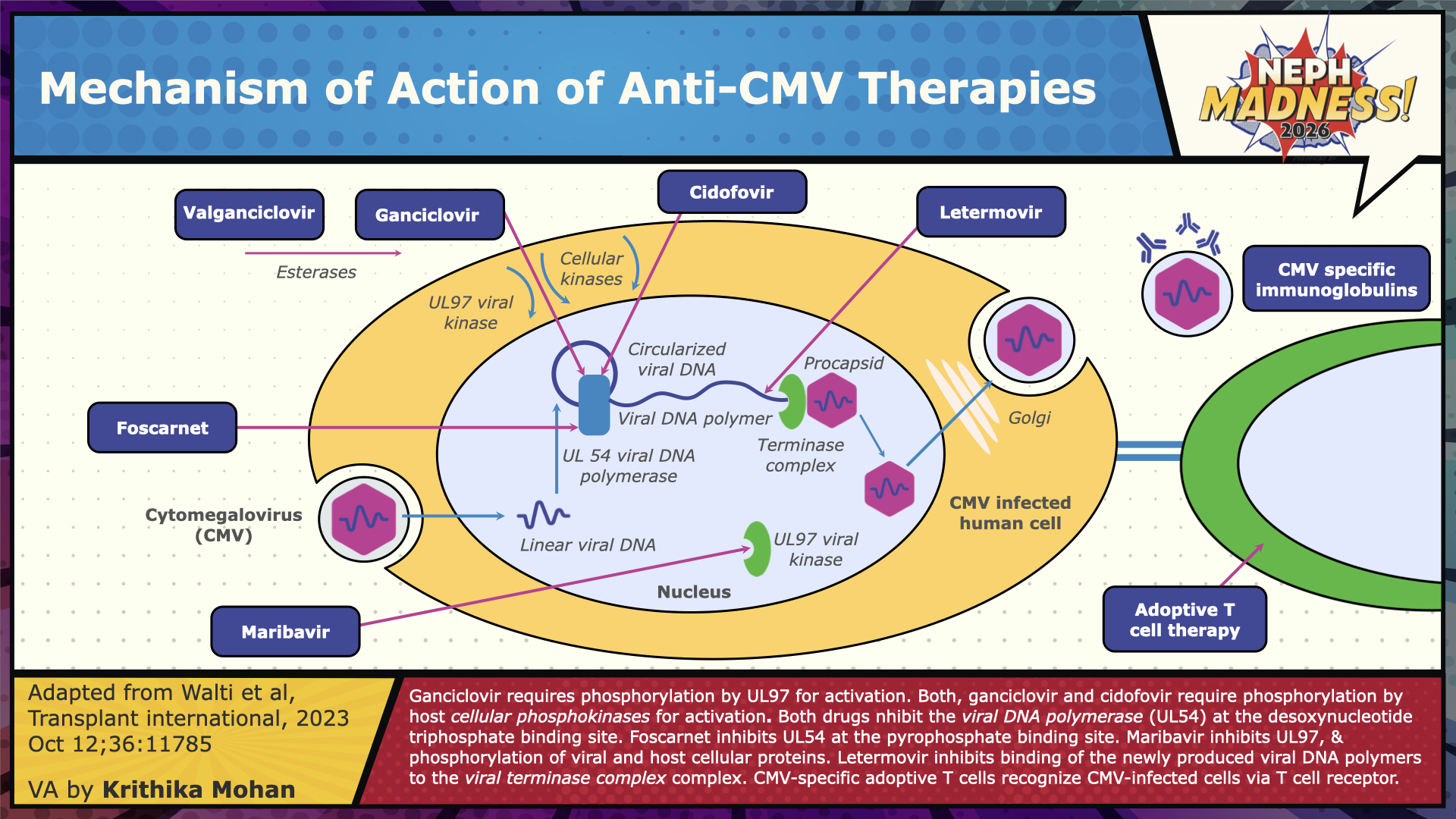
Refractory CMV infection and disease are suspected when there is a worsening of viral load and end-organ damage, respectively, despite 2 weeks of adequate antiviral treatment. This may be related to genotypic mutations in the virus that confer resistance to specific antiviral medications, which we refer to as “resistant CMV”, or to host factors such as high viral load at detection, profound immunosuppression, or insufficient drug levels at the site of infection. The management of refractory disease involves optimizing the dosing and route of Ganciclovir, reducing immunosuppression, performing resistance testing, and switching to second-line therapies. The anti-viral options available for empiric treatment of refractory CMV infections (with or without resistance [R/R]) include Foscarnet and Cidofovir, which are direct pUL54 inhibitors, and maribavir, which is a pUL97 inhibitor. Maribavir is recommended as the principal alternative for refractory/resistant disease by the Transplantation Society International CMV Consensus group, based on a recent study showing superior viremia clearance and symptom control, except for clinically unwell cases with high viral loads, in which case Foscarnet is suggested. Resistance testing, when available, can guide the choice of therapy. Letermovir monotherapy is not recommended for the treatment of active infections, as it lacks efficacy data and has a low genetic barrier to resistance development. CMV immunoglobulin (CMVIG) or pooled IVIG may be used as adjunctive therapy in the management of CMV disease, though the evidence for this practice is not compelling.

The Future: New Treatments and The Pipeline
Ongoing research focuses on methods to address the immune deficiencies, particularly the lack of CMV-specific T-cells, which are the driver of CMV infections after transplantation. Vaccination is an attractive approach, and several CMV vaccines have been studied or are currently under investigation. There is evidence supporting their safety; however, they have a varying degree of efficacy, and none have provided adequate protection so far, and hence are not approved for routine clinical use. One of the vaccine candidates, Triplex, elicited CMV-specific T-cell responses showing preliminary evidence of efficacy in Hematopoietic Cell Transplant, and is under active investigation in solid organ transplantation. Multiple anti-CMV monoclonal antibodies have been developed; however, none have been approved for clinical use yet.
Adoptive T-cell therapy is a promising approach. In this treatment, T-cells from the patient’s peripheral blood (autologous) or a donor (allogeneic) are extracted and expanded ex vivo by stimulating them with CMV antigens, then infused back into the patient. Many virus-specific T-cells (VSTs) are being studied and have demonstrated safety and effectiveness in treatment and prevention in Phase I/II trials, particularly in managing resistant or refractory CMV disease. Multivirus-specific T-cell therapy has emerged and is currently under evaluation, with the potential to significantly alter the treatment landscape.
With the recent advancements achieved and the ongoing efforts in managing CMV infection, we may be approaching a future where this troll is more definitively tamed. But until then, we must all keep our (owl) eyes out for this troll.
In this episode of Febrile Podcast, join Drs. Caitlyn Vlasschaert, Hariharasudan Natarajan, Sam Kant, Jeannina Smith, and Samira Farouk as they chat about the Trolls of Transplantation Region!
130: Trolls of Transplant with NephMadness 2026!
– Executive Team Members for this region: Samira Farouk @ssfarouk.bsky.social and Ana Catalina Alvarez-Elías @catochita – @catochita.bsky.social | Meet the Gamemakers
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC per region through NKF PERC (detailed instructions here). The CME and MOC activity will expire on June 1, 2026.
Leave a Reply