
Sickle Cell Disease: Peering Behind the Veil…
The association between sickle cell disease (SCD) and chronic kidney disease (CKD) is well known. A disease of recurrent vascular occlusion due to sickling red cells, the renal risk from such occlusive events is not to be understated. The kidney is highly susceptible to recurrent injury from such vaso-occlusion; the medulla, in particular, is more susceptible to damage, given the decreased oxygen tension at this level.
This type of injury manifests in a variety of different ways. Early on, the kidney can demonstrate compensatory changes with adaptive hyperfiltration. As the kidney endures repetitive injury, papillary necrosis and associated loss of concentrating capacity can occur, and – if our current understanding of the pathophysiologic injury pattern is correct – eventually progressive nephron loss and CKD can develop.
Historically, the life expectancy of those with SCD was so limited that long-term consequences of the disease were largely irrelevant. Over the last several decades, with progress in sickle cell treatment and steadily increasing life expectancy for those affected by the disease, long term sequelae are now more common.
As nephrologists, addressing the needs of these patients is challenging. It is incredibly easy, as in so many diseases with renal implications, to wave our hands and advise maximal management of the causative disease. Questions remain, however: What else do we know? How do we advise patients about what is coming? How can we find those patients most likely to benefit from more intensive therapies?
In a recent retrospective observational study published in AJKD on this topic, Derebail et al set out to look at what disease characteristics might inform our prognostication in patients with SCD. This isn’t the first study that has sought to define these risk factors but may be the largest with 427 patients. Patients included in the study were from a single center between 2004 and 2013. Mean age was 30 years, with an approximate 50/50 male/female split. Baseline data also included genotypes, which were used to stratify patients into either severe or mild genotypes:
SEVERE: homozygous SCD [HbSS] or sickle-β0-thalassemia
MILD: hemoglobin SC disease [HbSC] or sickle- β+-thalassemia [HbSβ+)
These genotypes correlate with phenotypic severity in patients with SCD. This separation is supported by observations within the selected cohort; patients with the severe genotype had higher markers of cell turnover (LDH, reticulocyte count, bilirubin, etc.) as well as higher rates of sickle events (acute chest syndrome, CVA, priapism, etc). Notably, starting creatinine between the two groups was comparable and overwhelmingly suggested compensatory hyperfiltration (median eGFR, calculated with CKD-EPI, for all patients was 134 mL/min/1.73 m2). Only approximately 20% of all patients had prevalent CKD, which was defined as an eGFR <90 mL/min/1.73 m2 or the presence of proteinuria. It was slightly more prevalent in the severe SCD genotypes than among those with mild disease.
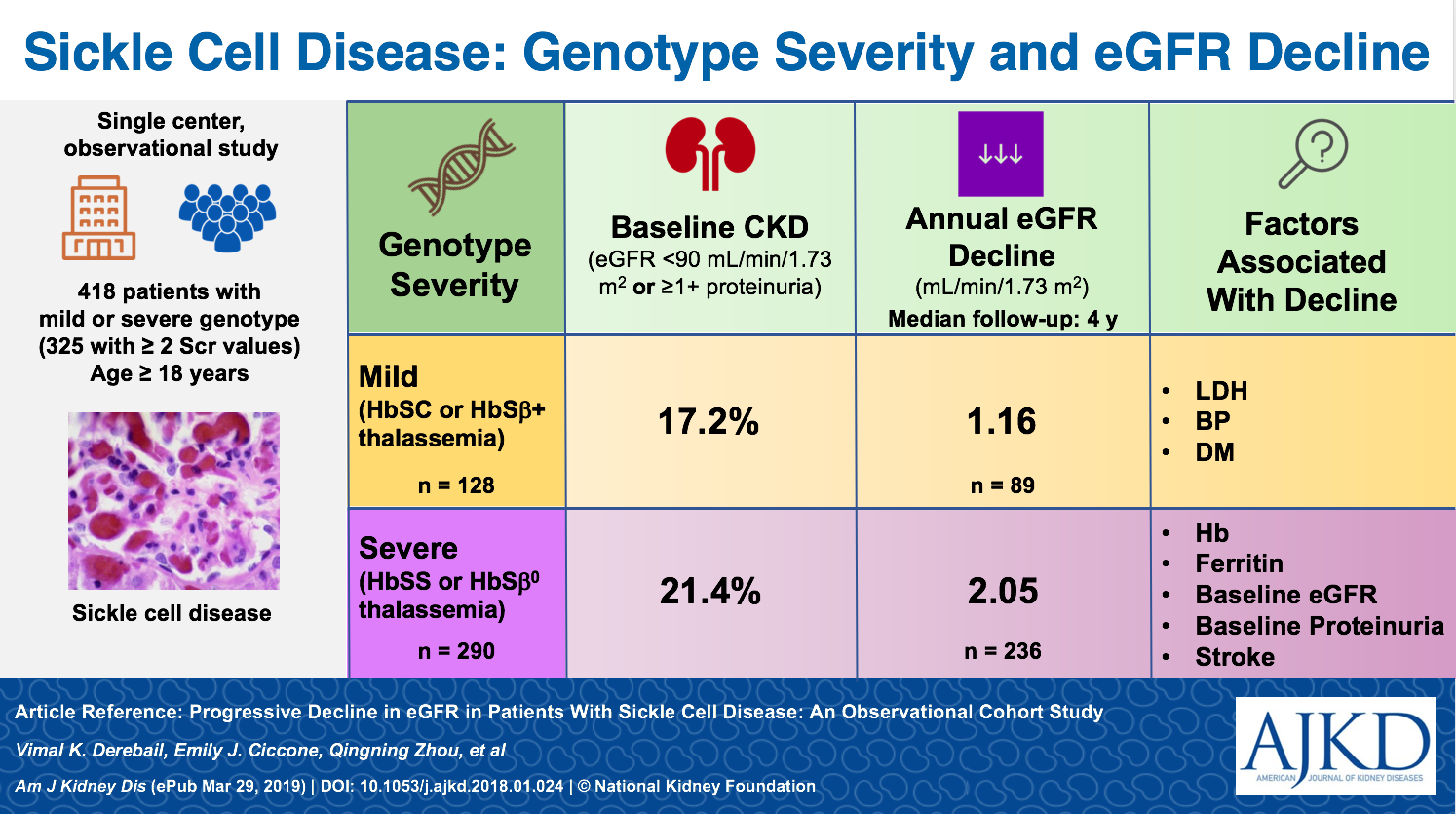
The median follow-up was four years for the cohort with approximately six creatinine measurements used to estimate the slope of eGFR decay. I don’t think it will come as any surprise that patients with more severe disease were found to have a slightly greater slope in eGFR decline. Severe genotypes started with slightly higher eGFR at enrollment – presumably due to worse hyperfiltration – and had eGFR decline of 2.05 mL/min/1.73 m2 per year. This compared to an average decline of 1.15 mL/min/1.73 m2 per year among patients with mild genotypes.
Slope analysis in small cohorts is fraught with risk, either to underestimate the rate of decline among patients with mild disease, or to be overwhelmed by patients who have more severe disease. In this case, that concern is mitigated through the genotypic stratification chosen by Derebail et al. Those patients with more severe disease, and therefore more frequent disease exacerbations, are captured as a separate entity.
While the observed separation did not reach statistical significance, it does support what we might expect based on our understanding of pathophysiologic injury patterns seen in SCD. This observed trend and the development of progressive CKD is also emboldened by the emerging body of literature around recurrent AKI events and CKD risk.

Slope of estimated glomerular filtration rate (eGFR) decline in individuals with sickle cell disease. Slope estimates also provided for subgroup with severe genotypes (homozygous sickle cell disease [HbSS] and HbSβ0-thalassemia) and mild genotypes (hemoglobin CD disease [HbSC] and HbSβ+-thalassemia). Figure 1 from Derebail et al, AJKD, © National Kidney Foundation.
Whether these agents may increase the risk of acute kidney injury (AKI) during hypovolemic episodes is another consideration. The use of such therapies was observed in the studied cohort; those patients with severe genotypes were more likely to be treated with ACEi/ARB therapy (OR 6.1). Was that use because of their observed CKD and the presumption that such treatment would mitigate the consequent injury, or did it exacerbate AKI events and enhance the association with progression? I lean towards the former, but I don’t think the data here can answer that question, which deserves a dedicated investigation.
The proliferation of epidemiologic data is useful insofar as it instructs us on what questions to dive into with additional trials. The clinical trialist deep inside me knows that the ability to recognize the differences among affected patients allows us to look at a specific question among patients likely to see the effect of the interest. Using information like this may help us to refine those questions and target cohorts; perhaps by selecting those patients most likely to be affected, we can reduce trial duration and size, thereby reducing cost. Recognizing those as our principle barriers, we can use information like this to best target future interventional studies.
All of that said, this work probably does not significantly alter my current practice. I probably have been describing this phenomenon to my SCD patients with CKD (admittedly few) since I started seeing patients. I can imagine myself telling them that their kidneys are “innocent bystanders” in their broader disease, and that recurrent episodes will weigh on their kidneys over time.
I don’t know that Derebail et al will significantly modify my messaging in clinic. That said, it does offer support to my statements. For hematologists, it may also shed light on what risk factors may be tracked to observe for emergent CKD, promoting early referral to nephrology. Further, I hope that this work offers a doorway for future work that may alter the way that I counsel my patients with SCD.
– Post prepared by Ed Gould, AJKDBlog Contributor. Follow him @paininthekidney.
To view Derebail et al (subscription required), please visit AJKD.org.
Title: Progressive Decline in Estimated GFR in Patients With Sickle Cell Disease: An Observational Cohort Study
Authors: V.K. Derebail, E.J. Ciccone, Q. Zhou, R.R. Kilgore, J. Cai, and K.I. Ataga
DOI: 10.1053/j.ajkd.2018.12.027
Leave a Reply