
#NephMadness 2026: C3G Region
Submit your picks! | @NephMadness | @nephmadness.bsky.social | NephMadness 2026
Selection Committee Member: Bradley Dixon
Dr. Dixon is currently Professor of Pediatrics and Medicine and chief of the Section of Pediatric Nephrology at the University of Colorado School of Medicine and Children’s Hospital Colorado. Dr. Dixon’s clinical interests focus on complement-mediated renal diseases such as Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. Dr. Dixon is also an investigator in several clinical trials of complement-targeted therapies in these diseases.
Writer: Aarushi Varshney
Aarushi Varshney is a second-year nephrology fellow at Duke University. She completed her internal medicine residency at the University of Central Florida/HCA Consortium. Her clinical interests include glomerular diseases and home dialysis modalities.


Competitors for the C3G Region
What a ride it’s been for C3G. Back in 2015, we were just getting used to the new name and updated classification, and now here we are in 2026 with actual approved treatments on the table. It’s amazing to see how something as complex and intimidating as the complement cascade is finally becoming more manageable. As nephrologists, the time is now to embrace the complement cascade, because it’s shaping the way we diagnose and treat these patients. And of course, kidney pathology remains at the center of it all, helping us connect the biology to what we actually see in the tissue. Who will it be, Team Diagnosis or Team Treatments?
Team 1: C3G Diagnosis
versus
Team 2: C3G Treatment
Image generated by Matthew Sparks using ChatGPT at http://chat.openai.com, February 2026. After using the tool to generate the image, Sparks and the NephMadness Executive Team reviewed and take full responsibility for the final graphic image.
In this episode, hosts Kenar Jhaveri and Koyal Jain are joined by Matt Sparks and Aarushi Varshney to discuss the evolving landscape of C3 Glomerulopathy (C3G). The conversation highlights the shift from traditional electron microscopy-based classifications to modern immunofluorescence-based diagnosis, as well as the groundbreaking arrival of two new FDA-approved targeted therapies.
Episode 11: Nephmadness Special! Matt Sparks and Aarushi Varshney on C3G
COMMENTARY BY ANUJA JAVA:
C3G Treatments Look Poised to Win it All, A Massive Victory for Patients
Team 1: C3G Diagnosis
Getting the right diagnosis is crucial to success. Strong C3 staining is not specific and can be mimicked by infection‑related GN, paraprotein‑associated disease, and other complement‑activating processes. Careful clinicopathologic correlation, including evaluation for infections and monoclonal proteins, is essential to avoid mislabeling these look‑alike entities as true C3G. Team Diagnosis in C3G is the frontrunner because accurate diagnosis is the critical first step that determines the right pathway, the right therapy, and the right outcome.

Copyright: Dudarev Mikhail/ Shutterstock
C3 glomerulopathy (C3G) is an ultra-rare disorder that encompasses C3 glomerulonephritis (C3GN) and dense-deposit disease (DDD). C3G results from dysregulation of the alternative complement pathway, leading to C3 dominant complement deposition in the kidney. The estimated incidence of C3G ranges from 1 in 300,000 to 1 in a million individuals. Even though kidney biopsy is the main diagnostic tool, it’s not straightforward when it comes to diagnosing C3G due to several nuances which makes it rather challenging for both nephrologists and pathologists to diagnose. This ultra-rare entity sits at the intersection of histologic overlap, subtle immunofluorescent interpretation, inadequate complement testing, and evolving terminology.
First, beginning with the former nomenclature of membranoproliferative or mesangiocapillary glomerulonephritis (MPGN), the traditional classification categorized glomerular diseases primarily based on the findings of light microscopy (LM) and electron microscopy (EM) of kidney tissue.
The findings were divided into three categories depending on the type of deposits seen in the kidney biopsy. MPGN type II was characterized by C3 deposits within the mesangium along the glomerular basement membrane (GBM) surfaces, whereas MPGN type I and III (also referred to as immune complex [IC-MPGN]) contained monoclonal or polyclonal immunoglobulin deposits. Thus, if an IC-MPGN pattern of injury is observed, patients should be screened for occult infection or in the case of adults, paraproteinemia with serum protein electrophoresis (SPEP) and immunofixation electrophoresis (IFE).
Advances in research and expert consensus reshaped our understanding of these entities, leading to the establishment of the new umbrella term C3G in 2013. Deposition of C3 along with absence or near-absence of immunoglobulin deposition is the classic diagnostic criteria for C3G. Differentiating between the subtypes of C3G entities can be challenging, as overlapping features are common.
The diagnosis can be further complicated by variability of pathology interpretation, especially when it comes to grading the immunofluorescence (IF) staining intensity. Subtle differences in staining intensity may shift a case toward or away from being classified as C3G. Before diving into IF, let’s try to differentiate DDD from C3GN. The distinction relies primarily on EM findings. In DDD, dense “sausage-shaped” or “ribbon-shaped” deposits cause marked thickening of the GBM and may even extend to Bowman’s capsule and occasionally to the tubular basement membrane. In contrast, C3GN deposits are not as prominent as DDD, and they demonstrate cloudy, amorphous dense material mainly within the mesangial and subendothelial areas. Both subtypes can have subepithelial deposits. Additionally, DDD is the less common form of C3G, and can present with extra-kidney features, including ocular drusen and partial lipodystrophy.
Returning to the IF topic, using a strict criterion of C3-only staining identified only 50% of DDD cases, whereas applying a broader definition (C3 staining at least two orders of magnitude greater than immunoglobulin) identified about 88% of DDD cases. Based on this variability, a consensus definition of C3 dominance by at least two orders of magnitude was adopted as the standard criteria for diagnosing C3G. However, these C3-dominant staining findings on immunofluorescence are not objective —rather, subjective and semiquantitative —which can make the diagnosis even more challenging, since it depends on the observer’s interpretation.

Another diagnostic dilemma arises when post-infection associated glomerulonephritis (PIGN) has C3-dominant deposits, closely mimicking C3G. Typically, PIGN should have recovery of kidney function to the baseline within a few weeks. However, if hematuria and proteinuria persist beyond 12 weeks and the biopsy is consistent with C3 dominance, then the diagnosis should shift towards C3G, warranting workup for alternative complement pathway dysregulation, genetics, and autoantibodies (anti complement factor H, C3/C4/C5 nephritic factors).
Enough with challenges, let’s discuss the tools which can help us “rule in” C3G. As stated before, C3G occurs due to dysregulation of the alternative complement pathway (one of the three proximal complement pathways- classical, lectin, and alternative pathway). As shown below, the alternative complement pathway is constantly active due to spontaneous hydrolysis of the reactive thioester bond in C3, known as “tick-over”. Once initiated, C3 is cleaved by C3 convertases, C4b2a (classical/lectin) and C3bBb (alternative) into C3a and C3b. C3b binds to factor B, which is cleaved by factor D to form C3bBb leading to amplification of the response. As C3b accumulates and C5 convertases form, this leads to a formation of membrane attack formation (C5b-9) and cell lysis.
If formed autoantibodies cause the C3bBb complex to remain active or block normal inhibitory mechanisms, this dysregulation of the complement pathway can lead to C3G. The primary driver is C3b accumulation, which is why C5 inhibitors generally are less effective than therapies targeting more proximal nodes in the complement pathway.

In cases where C3G is suspected, complement testing for autoantibodies can help further solidify the diagnosis. The figure shown below presents an overview of cohort-averaged percentages for common complement abnormalities (both genetic variant and autoantibodies). The graph illustrates that autoantibody-mediated mechanisms are more frequent; however, it is important to highlight that negative genetic testing or negative autoantibody testing does not rule out an underlying complement abnormality which can still give rise to C3G pathology.
When it comes to a workup that is cost-friendly and widely available, C3 and C4 testing is a reasonable starting point. Some patients with C3GN will have a low serum C3 level which is more commonly seen in DDD. Defining a “low” serum C3 cutoff for C3G workup is challenging. Recent papers and trials have used thresholds such as <77 mg/dL (or <0.85 * lower limit of the normal range) and even <90 mg/dL; however, the exact cutoff remains uncertain. Also, an infectious workup plus SPEP and IFE testing is a good starting point to rule out other etiologies.
A small subset of patients may also have low serum C4 levels, although this is uncommon. That being said, these labs are supportive at best and many patients with C3G can still have normal C3 and C4 levels, so normal values do not rule out the diagnosis.
When you move on to more specialized testing such as factor H autoantibodies, C3/C4/C5 nephritic factor, or factor B autoantibodies, these abnormalities can be seen in some patients with C3G, again more common in DDD than C3GN. However, these results can vary and are not always seen, so they help support the diagnosis, but are not definitive on their own. As is often the case when it comes to genetic testing, the results may reveal variants in complement genes but many detect variants which have uncertain pathogenicity.
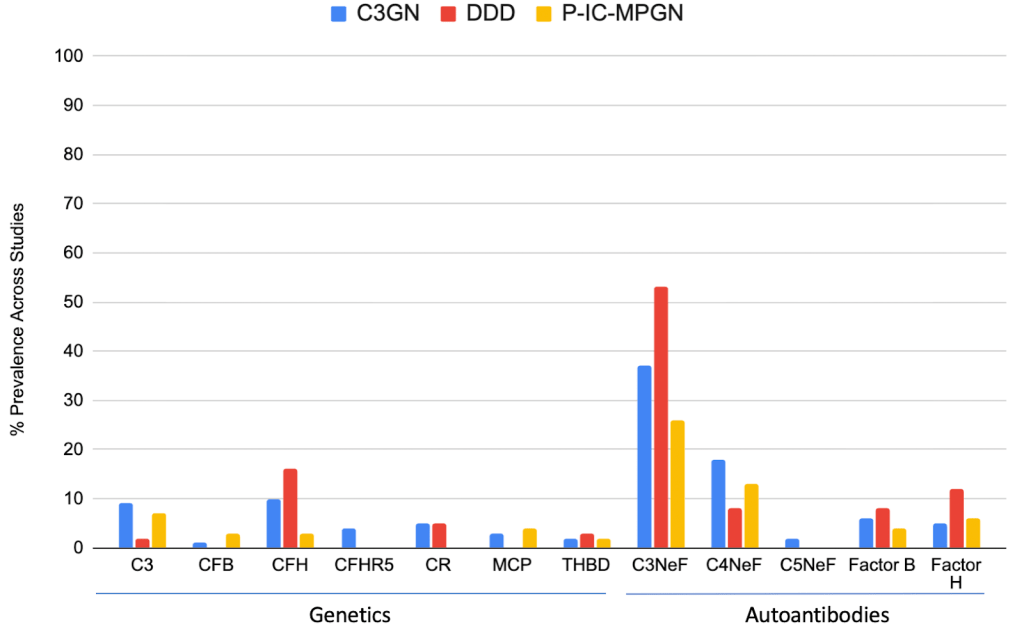
Figure adapted from Bomback et al 2024 KI Reports showing prevalence of both genetic and acquired autoantibodies in C3GN, DDD, and P-IC-MPGN.
Team 2: C3G Treatment
Team Treatment of C3G is rising fast, leaving behind the disappointing era of steroids and MMF, which rarely altered the disease’s trajectory. Novel complement‑targeted therapeutics like pegcetacoplan and iptacopan are finally addressing the root biology of C3G with precision rather than blunt immunosuppression. With these agents leading a rapidly expanding pipeline, this team embodies real optimism for durable remission and a brighter future for patients.

Copyright: Wollertz/ Shutterstock
For a long time, management of C3G was limited and relied on untargeted immunosuppression such as steroids and mycophenolate mofetil. Despite treatment, approximately 30 to 50% of patients progress to kidney failure within 10 years of their diagnosis. Unfortunately, even after successful management of kidney transplantation, C3G has a high risk of recurrence. However, we now have an opportunity to reshape the trajectory of C3G.
Though guidelines are still in the process of being written for C3G management, there has been a paradigm shift in the treatment of C3G in the form of more proximal inhibitors of the alternative complement pathway (Factor B, C3b, and C3 inhibitors). These therapies have ushered in a new era in the treatment of C3G and are reshaping the treatment of this ultra-rare disease and hopefully will result in a dramatic reduction in kidney failure.
Let’s start off with the two new kids on the block for C3GN treatment: iptacopan and pegcetacoplan.
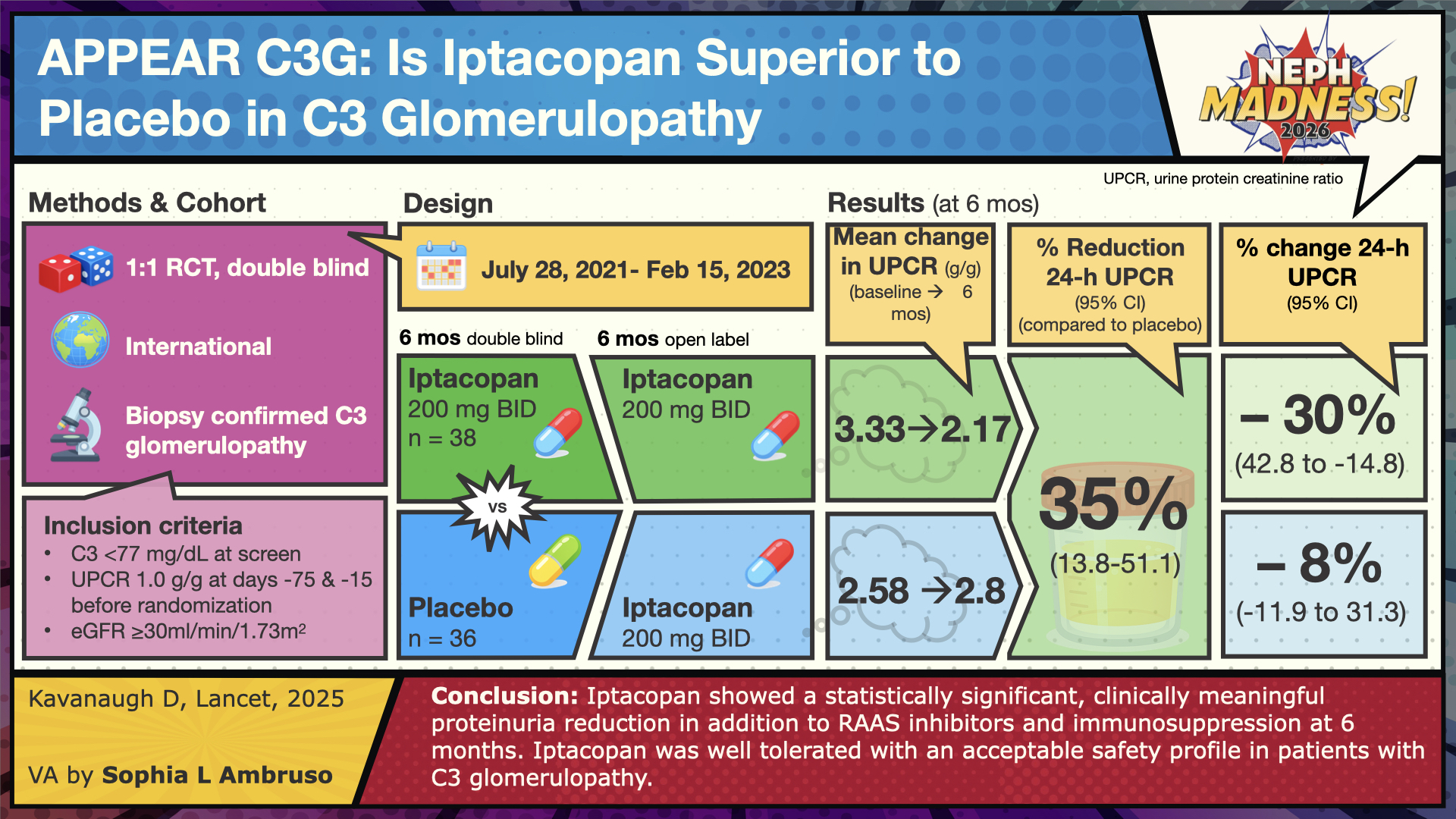
APPEAR-C3G was a Phase 3 trial evaluating iptacopan, an oral complement inhibitor that targets factor B. By inhibiting factor B, iptacopan disrupts C3 convertase activity, thereby preventing C3 activation and blocking the alternative pathway amplification loop. Participants were 18 to 60 years old. One treatment group (n=38) received iptacopan 200 mg twice daily (BID), while the placebo group (n=36) received matching placebo capsules BID for 6 months, followed by an open-label extension with iptacopan 200 mg BID for an additional 6 months. Key inclusion criteria included: (1) reduced serum C3 level (<77 mg/dL) at screening, (2) urine protein-to-creatinine ratio (UPCR) ≥1000 mg/g, (3) estimated glomerular filtration rate (eGFR) ≥30 mL/min/1.73 m², and (4) vaccination against Neisseria meningitidis and Streptococcus pneumoniae. Exclusion criteria included prior cell or solid organ transplantation, rapidly progressive crescentic glomerulonephritis (RPGN), and monoclonal gammopathy of undetermined significance (MGUS). Baseline demographics showed that most participants in both arms were Caucasian. Median baseline 24-hour UPCR was 3000 mg/g in the iptacopan group and 2600 mg/g in the placebo group, and approximately 70% of patients in each arm had an eGFR >60 mL/min/1.73 m².
Kidney biopsy findings revealed that the predominant C3G subtype was C3GN, present in approximately 68% of patients in the iptacopan group and 89% in the placebo group. DDD accounted for 24% (9 of 38) of patients receiving iptacopan, compared with only 3% (1 of 36) in the placebo arm.
Both groups received maximally tolerated supportive therapy with ACE inhibitors or ARBs. The protocol permitted continuation of additional agents, including mycophenolic acid, low-dose corticosteroids (<7.5 mg/day), SGLT2 inhibitors, and mineralocorticoid receptor antagonists. Baseline demographics showed an average age in the mid-to-late 20s, with most participants having preserved kidney function (eGFR ≥90 mL/min/1.73 m²). Approximately 60–80% of patients in each arm had the C3GN subtype of C3G.
The primary endpoint was proteinuria reduction at 6 months. The secondary outcome was to evaluate for differences in eGFR from baseline at 6 months along with several other endpoints (patient-reported fatigue, reducing glomerular inflammation in the kidney along with evaluating safety-tolerability of iptacopan).
The results showed that treatment with iptacopan versus placebo resulted in a 35% relative reduction in proteinuria (p=0.0014) at 6 months, sustained through 12 months (open-labeled period). For the secondary outcome, the change in eGFR at 6 months did not differ significantly between groups (p=0.3241); eGFR remained stable in both groups. For changes in other variables (serum C3, plasma sC5b-9 aka MAC complex, urinary sC5b-9:creatinine), the results favored iptacopan over placebo. On the histologic activity scale, iptacopan had a statistically significant effect on two components (endocapillary proliferation and leukocyte infiltration) out of the six parameters evaluated. Also, patient-reported fatigue scores did not differ amongst both groups. The medication was well tolerated. No deaths occurred. In terms of severe adverse effects (SAE), 8% (N=3) of participants in the iptacopan group had an SAE (blood cultures positive for streptococcus pneumonia, infected bite, or chest discomfort) compared to 3% (N=1) in the placebo group (acute kidney injury or ascites).
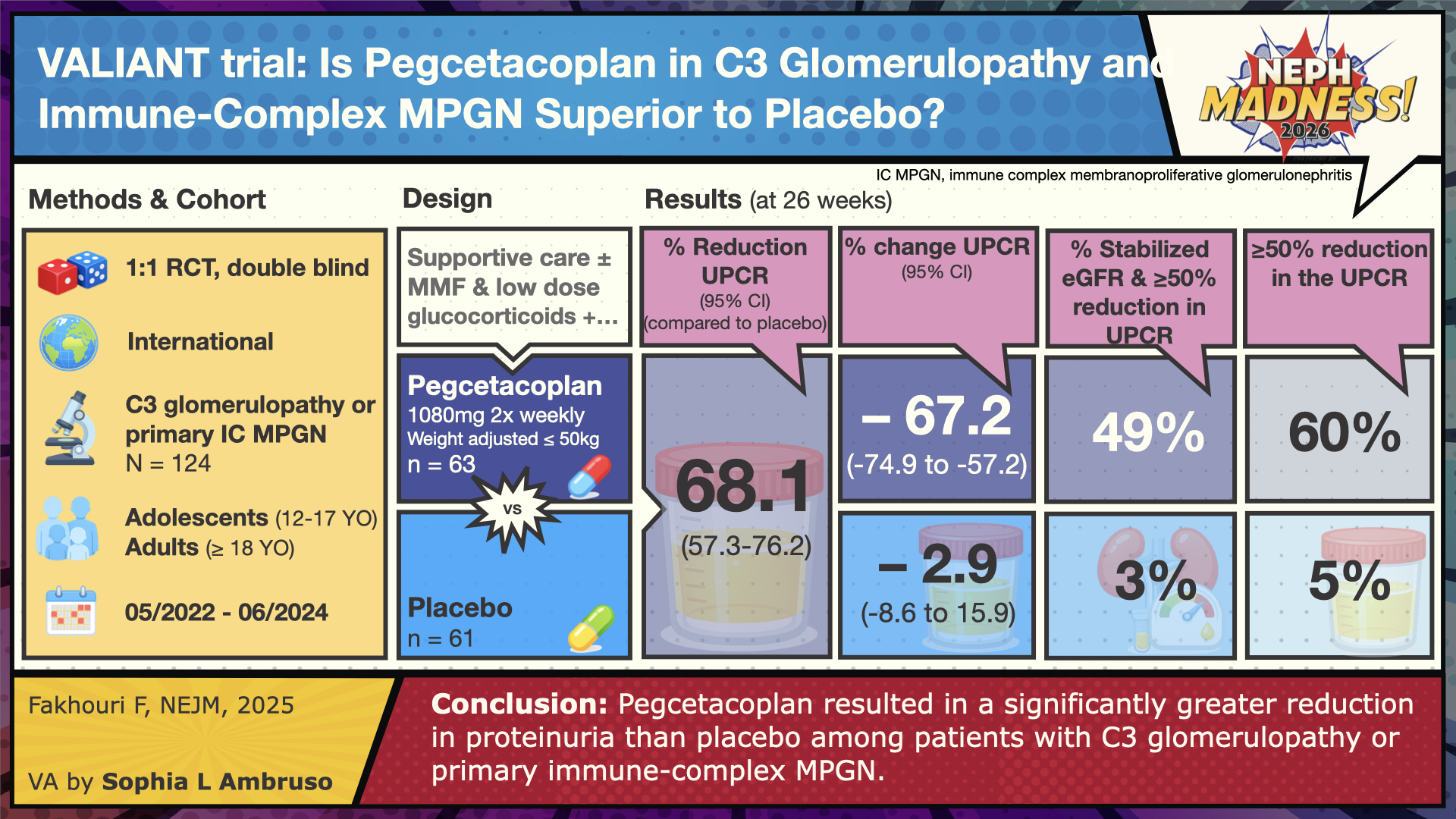
The VALIANT phase 3 trial published in December 2025 looked at pegcetacoplan use (given via subcutaneous infusion twice a week at home) in patients with C3G and primary IC-MPGN. Unlike the APPEAR-C3G trial, this study also included kidney transplant recipients with recurrence of disease, which is common and associated with graft loss in up to 60% of cases. Pegcetacoplan binds directly to C3 and C3b, inhibiting their cleavage and downstream amplification. As a result, it blocks activation of all three complement pathways: classical, lectin, and alternative. VALIANT had broader inclusion criteria compared to APPEAR-C3G; adolescents aged 12-17 years were included and made up approximately 56% of the study in both the treatment and placebo group; adults accounted for about 44% in each group. On kidney biopsy, the majority of the patients were found to have C3G findings (81% in pegcetacoplan and 74% in the placebo group), whereas roughly 20% had primary IC-MPGN pattern of injury. A small subgroup had a history of kidney transplantation: five patients in pegcetacoplan and four in placebo.
Similar to APPEAR-C3G, patients received maximally tolerated supportive therapy and background immunosuppression was continued if already in use. The median UPCR was ~2,400 mg/g in the pegcetacoplan arm (n=63) and 1800 mg/g in the placebo arm (n=61) with eGFR >60. The primary outcome was UPCR reduction in 26 weeks. Secondary outcomes included eGFR reduction with delta change, at least 50% reduction in UPCR, and activity scoring of C3 in histology along with decrease in staining of C3. The exciting part of this trial was really in the results section. The treatment resulted in a 68.3% reduction in proteinuria, along with significant reduction in C3c staining (74% compared to 12% in placebo) and stabilized eGFR finding.
The adverse events were similar in both groups, but one patient in pegcetacoplan died from acute respiratory failure associated with COVID-19 pneumonia. Importantly, there were no cases of kidney allograft rejection or failure.
Both medications have shown promising results. Pegcetacoplan showed greater reduction in proteinuria compared with iptacopan, though it requires twice a week injection, while iptacopan (being oral) can be a more convenient and noninvasive regimen.
Because both therapies inhibit the complement cascade—a key defense against encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae— patients should be vaccinated against these pathogens before starting treatment. The long-term infection risk with complement-targeted therapies remains uncertain. When urgent initiation is necessary and the vaccination series cannot be completed (minimum 2 weeks), antibiotic prophylaxis is advised. Recommended options include Penicillin V or amoxicillin; for penicillin-allergic patients, alternatives are ciprofloxacin or azithromycin.
Now that we have covered therapies with proven benefits, let’s shift to other therapies where evidence is less clear cut, starting off with the C5a receptor blocker avacopan. The ACCOLADE phase 2 trial evaluated avacopan, in 57 patients with biopsy-proven C3G over 26 and 52 weeks. The trial evaluated both adults (18-80 years of age) and adolescents (12-17 years of age) and included patients with kidney transplants. The primary end point was to evaluate the histological changes from baseline to week 26. The secondary end points included change in UPCR and eGFR at week 26 compared with the placebo group. The primary end point, change in the C3G histological activity index, was not statistically significant compared to the placebo group. Though the secondary endpoints of eGFR and UPCR showed modest improvement they were not statistically significant at week 26. The medication was overall well tolerated without serious adverse events. Thus, avacopan is not recommended in C3G.
Given the underlying pathogenesis of C3G and the findings from the APPEAR-C3G and VALIANT phase 3 trials, it is unsurprising that avacopan failed to show significant benefit. Avacopan targets the distal complement pathway, whereas C3G is primarily driven by dysregulation of the proximal alternative pathway. Supportive therapy with RAS inhibitors and SGLT2 inhibitors should be used, while noting that direct evidence is limited. This ultra-rare disease may have been included within the glomerular subgroup analysis in both the DAKA-CKD and EMPA-Kidney studies, but the trials excluded patients on or recent exposure to immunosuppression. However, in cases of nephrotic range proteinuria or rapid kidney function decline, management becomes more complex.
Currently, there is no definitive evidence supporting the efficacy of plasma exchange in C3G. Although this intervention can transiently reduce circulating autoantibodies, these typically re-emerge following discontinuation, often resulting in recurrent kidney injury. Consequently, plasma exchange should be considered a short-term, temporizing measure rather than a durable therapeutic strategy. Further studies are warranted to delineate its potential role within the broader management framework of C3G. What about rituximab? Rituximab targets CD20 expressing B-cells, leading to B-cell lysis. Prior studies have shown a promising response in immunoglobulin-associated MPGN; however, in regards to complement-associated C3GN and DDD, its efficacy has been less consistent.
Shifting focus to general immunosuppressive therapies —which are aimed at dampening the immune response in C3G — treatment with mycophenolate mofetil and corticosteroids have shown inconsistent results across multiple studies, none of which were randomized controlled trials, with no clear evidence supporting its efficacy.
Given the strong efficacy demonstrated by iptacopan and pegcetacoplan, terminal complement inhibition (eculizumab/ravulizumab) will likely be reserved for patients who are intolerant of or unresponsive to these agents. The therapeutic focus has shifted toward strategies that specifically target the dysregulated alternative complement pathway in affected individuals. Several case reports and series have examined the use of terminal complement inhibitors such as anti-C5 therapies like eculizumab/ravulizumab in patients with C3G, but the evidence remains limited and largely observational. Bombak et al. conducted one of the earlier studies involving patients with DDD and C3GN (n=5, both groups) and observed that eculizumab was beneficial in select cases. This was noted by reduction in proteinuria, improvement in serum creatinine after 12 months, and normalization of elevated membrane attack complex (MAC) (C5b-9) levels. Additional data from Le Quintrec et al. suggest that eculizumab may provide some benefit in approximately 50% patients with C3G, particularly in rapidly progressive cases, with a median treatment duration of approximately 14 months. However, evidence remains limited, especially in those with advanced kidney disease.
– Executive Team Members for this region: Matt Sparks @Nephro_Sparks – @nephrosparks.bsky.social and Anna Vinnikova @KidneyWars – @kidneywars.bsky.social| Meet the Gamemakers
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC per region through NKF PERC (detailed instructions here). The CME and MOC activity will expire on June 1, 2026.
Leave a Reply