
#NephMadness 2019: Pain Region
Submit your picks! | NephMadness 2019 | #NephMadness | #PainRegion
Selection Committee Member: David Juurlink
David Juurlink is a staff internist and head of the Division of Clinical Pharmacology and Toxicology (DCPT) at Sunnybrook. He is professor and head of the DCPT at the University of Toronto. He is also a medical toxicologist at the Ontario Poison Centre and a senior scientist at the ICES, where he maintains an active drug safety research program. Follow him @DavidJuurlink.
Writer: Samantha Gelfand @SammyG
Samantha Gelfand is a nephrology fellow at the University of Pennsylvania. Her academic focus is renal supportive care, including pain management, dialysis decision-making, and advance care planning. She will be doing a fellowship in Hospice and Palliative Medicine at Dana Farber-MGH after completion of nephrology fellowship at Penn.
Competitors for the Pain Region
Opioids vs NSAIDs
GABAnergics vs Tramadol

Copyright: Sherry Yates Young / Shutterstock
Ah, pain management, the Achilles heel of modern medicine. The struggle is real, and the tools are imperfect. While there are plenty of data now on the pandemicity of pain in the kidney disease patient population, treatment remains a bugaboo – there is no Kt/V for pain control adequacy, and the risks of pharmacologic treatment are significant.
If pushed to summarize, pain management in patients with kidney disease often goes something like this: NSAIDs are evil, acetaminophen hardly works, opioids are dangerous, and all the rest (tramadol, gabapentinoids, antidepressants) are messy.
Nonetheless, in a field in which cures are few, illness is chronic, and prognosis often worse than cancer, reduction of pain should be one of our highest aspirations. Whenever we consider giving a medication for pain, we must weigh the potential benefits against the potential harms. For this reason, we offer a match-up of our most common pharmacologic tools for pain management. We have presented each drug class separately, though OF COURSE, they should be used synergistically AND alongside the (elusive, evidence-based, and infuriatingly under-covered) nonpharmacologic tools for pain management.
{kind=link}
Opioids vs NSAIDs
Two teams of troublemakers. Almost always picked last in any draft, the players are imperfect and notorious for personal fouls.
Opioids
The analgesic property of this group was originally discovered in the naturally occurring alkaloids from the opium poppy, Papaver somniferum (think Dorothy + Co’s big nap outside the gates of Oz). All opioids bind with one or more opioid receptors (mu, kappa, delta), with analgesic properties dictated by their different patterns of binding (pure agonist, partial agonist, or mixed agonist/antagonist). The mu receptor is the main pain modulator, densest in the CNS but also present throughout the body.

Copyright: Pavel_Klimenko / Shutterstock
For the nephrologist, one of the key steps in prescribing opioids safely is knowing the metabolites, and specifically, whether they are 1) biologically active and 2) cleared by the kidney. Naturally occurring opioids (morphine, codeine) are to be avoided in patients with decreased kidney function due to their metabolism to morphine-3-glucuronide (M3G) and M6G. Both are cleared by the kidney, but only M6G is biologically active – it is a more potent mu agonist than its parent compound. It is thought to be responsible for morphine-related neurotoxicity in patients with diminished kidney function. To add insult to injury, though morphine and codeine are minimally protein-bound and moderately water soluble (and therefore readily dialyzable), M6G equilibrates slowly across the central nervous system (CNS); the basketball analogy here is actually too easy – even if the morphine “shot” is blocked (heroically, in the middle of the night, by naloxone or dialysis), there is a literal “rebound” over the subsequent hours, in which signs of toxicity can recur as the serum concentration increases after dialysis.
The semisynthetic opioids (hydrocodone, hydromorphone, oxycodone) and synthetic opioids (methadone, fentanyl) are more reasonable players. They are VERY powerful but can be coached not to overshoot. Hydromorphone, for example, is metabolized to H6G, which is cleared by the kidney but does not have biological activity — no analgesia, no known toxicity. It is inert. In a small study of 12 patients on dialysis with chronic pain on stable doses of hydromorphone, intra- and inter-dialytic concentrations of the drug and its metabolite were measured, confirming that hydromorphone does not accumulate in patients with anuria, and H6G is readily cleared by hemodialysis. Hence, the central position of hydromorphone in the “recommended” category in the WHO Preferred Analgesic Medications for Chronic Pain Management in CKD 4-5
The good news is that this line-up has a winning record. In a study (unfortunately small) in 2006, Barakzoy and Moss showed that among 45 patients on hemodialysis, 75% of whom had severe pain, use of this analgesic ladder for four weeks resulted in adequate analgesia in 96%. At the end of the study, 24% were in no pain, 71% were in mild pain, and 4% were in moderate pain. Interestingly, although the study did follow the WHO escalation ladder, the choice of agents were not necessarily biologically optimal. The agents used included gabapentin (38%), hydrocodone (27%), tramadol (24%), oxycodone (20%), nortriptyline (16%), and propoxyphene (2% — in one patient who had used it before and had requested it). Adverse effects were observed in three people. Patients reported more restful sleep, improved function, improved exertional tolerance, and better ability to tolerate dialysis.

Visual Abstract by @Errantnephron on Barakzoy et al
The bad news is that this adapted WHO ladder has several issues:
- Treatment decisions based on severity alone will land a whole lot of people on opioids, which may not be the most effective option, especially for chronic pain. Treatment decisions should be primarily based on the pain syndrome, characterized by cause (musculoskeletal, ischemic, mechanical, chemical), type (nociceptive, neuropathic, existential), chronicity (acute, subacute, chronic), and other features (allodynia, hyperalgesia, depression, insomnia). These aspects of the pain syndrome, in addition to consideration of potential harms, should determine the order in which different agents should be tried. In pain management, it is always a balance of potential harms and potential benefits. None of this is captured by this ladder. The CDC guidelines on use of opioids in chronic pain syndromes may be a better playbook, though still unable to do the heavy lifting of actual risk/benefit assessment for each individual patient.
- Buprenorphine and methadone prescriptions are restricted in the United States.
- There is a major reluctance among some clinicians to prescribe hydromorphone. There are many reasons for this – inexperience, experience, fear, aversion stemming from the common experience of being asked by patients for this med by name (“it’s the only thing that works”), or watching it sedate someone to the point of respiratory depression when inappropriately high doses are given. But this aversion leads to prescription of nonrecommended meds, including less potent opioids with more troublesome pharmacokinetics in patients with reduced kidney function (tramadol, oxycodone) or NSAIDs (toradol), whose toxicities can be worse than the imagined evils of vitamin D(ilaudid). This is borne out in the literature, as described below.
The elephant in the room, of course, is the opioid epidemic. At this point, few physicians believe that long-term use of opioids for nonmalignant pain is a great idea. Analysis after analysis has documented the high rates of dose-dependent harm and death resulting from long-term opioid use and addiction in the general population. Among patients with ESKD on hemodialysis, there is emerging evidence that confirms high rates of opioid prescription and higher mortality among those with a long-term prescription.
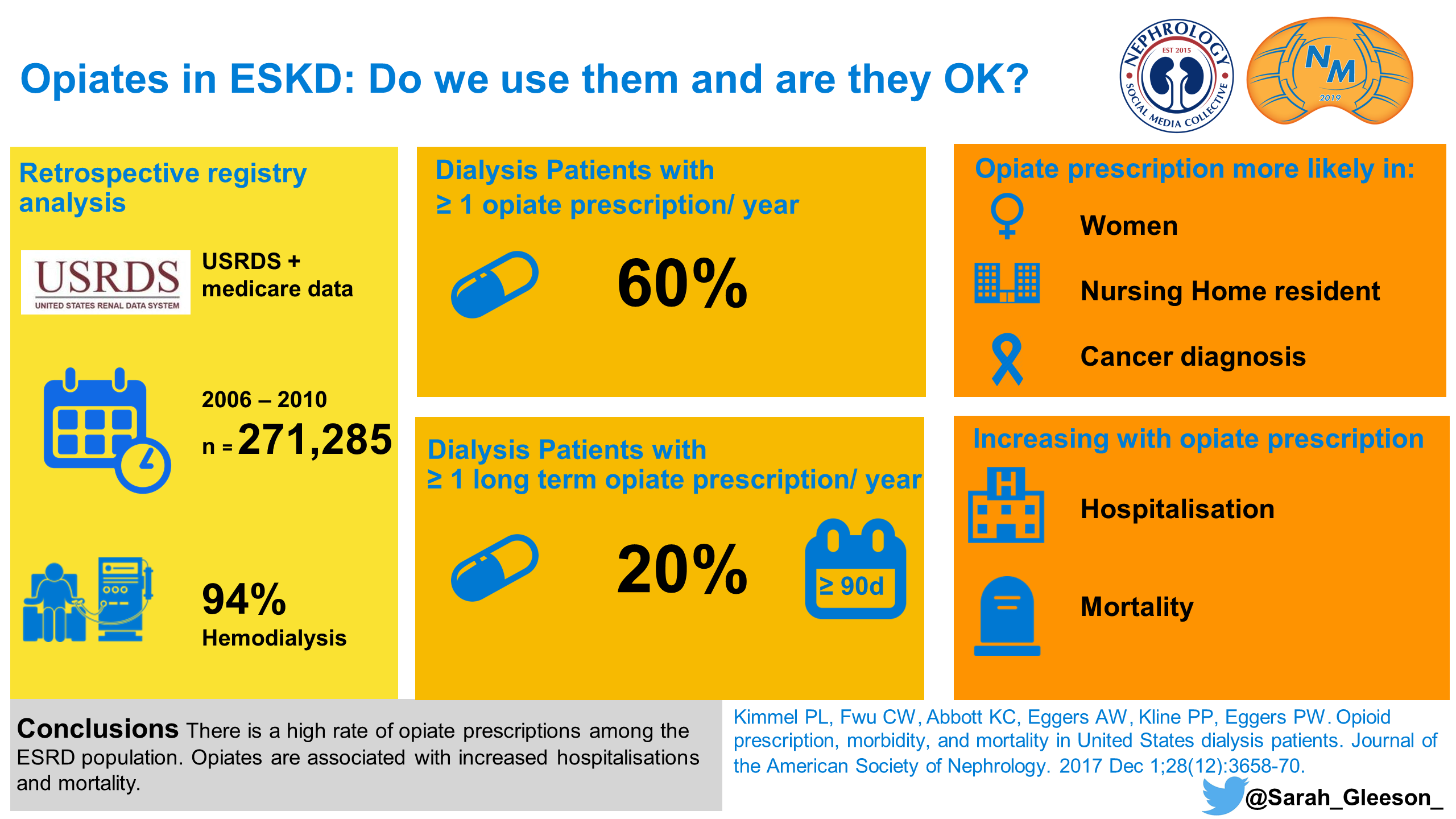
Visual Abstract by @sarah_gleeson_ on Kimmel et al
In another recent study, altered mental status (AMS), falls, and fractures are found to be more common in hemodialysis patients on opioids than hemodialysis patients not on opioids, although the overall increased relative risk was modest. These risks were higher among new opioid users and non-black patients. This is not terribly surprising. The more useful part of the study, however, was the comparison of hazard ratios by opioid agent, which confirmed the physiology-based recommendations of the WHO ladder — indeed, some opioids seem to cause way more problems than others. Spoiler alert: events were highest with codeine (HRs of 3.6 for AMS, 1.5 for fall, and 2.9 for fracture) and tramadol (HRs of 1.4 for AMS, 1.4 for fall, and 1.6 for fracture), though tramadol was the most commonly prescribed opioid.
What does this mean? It means that patients on codeine were 3.6 times more likely than people not on an opioid to have an episode of AMS. Events in the hydromorphone group, on the other hand, occurred less than in the codeine and tramadol groups (hydromorphone HR of 1.3 for AMS, 1.0 for fall, and 1.0 for fracture). This lower event rate is likely due to a number of factors, including the inert nature of hydromorphone main metabolite and its more predictable pharmacokinetics. All in all, there is an unspoken piece of advice here — if you are going to use opioids in hemodialysis patients, despite the known risks, choose hydromorphone over hydrocodone, oxycodone, or tramadol.
Unfortunately, the set-up of the study (which did not assess adequacy of pain control or quality of life) did not allow for any discussion of whether the demonstrated relative risks might be worth the benefit of pain control. And there’s the rub — risks are much more easy to measure, and worry about, than benefit, which depends on subjective and functional improvement. Additionally, to be fair, the authors do acknowledge that causal relationships cannot be inferred in these types of studies; there is always the potential for confounding by indication — that is, opioid prescription may be an illness marker. Some researchers have gone as far as to try to evaluate pain itself as the risk factor for poor outcomes.
NSAIDs
OK, this team has a tough track record. Despite being powerful tools against inflammatory nociceptive pain, the cyclooxygenase (COX) inhibitors have earned a bad reputation in the nephrologist’s world. As we all know (from the thousands of times we say it everyday on rounds and in notes), they are generally avoided in chronic kidney disease (CKD) and people at high risk for acute kidney injury (AKI).
A quick mechanistic review – they work by COX inhibition, which reduces prostaglandin synthesis from arachidonic acid. Less prostaglandin, less inflammation, less pain. Yay. However, in certain people, glomerular filtration rate (GFR) is dependent on prostaglandins (mostly E2 and I2) and can take a nosedive when NSAIDs come on board. Which people? Patients with kidneys that are already under other ischemic stressors (i.e., conditions of reduced effective arterial blood volume such as heart failure, cirrhosis, nephrotic syndrome). It also may occur in patients on other medications such as renin angiotensin system (RAS) blockers or those with volume depletion (i.e., diarrhea, vomiting, or acute blood loss). Prostaglandin-mediated afferent arteriolar vasodilation is a must in these individuals; without it, the kidney vascular supply is inadequate – the tubules can wither like an under-watered plant. To add insult to injury, the well-meaning juxtaglomerular apparatus flips out, mistaking the weak kidney perfusion for panic-level hypovolemia and goes into lost-in-the-desert conservation mode, gets real thrifty with sodium and potassium handling, and results in hypertension, edema, and hyperkalemia.

Copyright: Vladyslav Lehir / Shutterstock
These biochemical and hemodynamic effects aside, NSAIDs also are notorious for triggering interstitial and glomerular reactions in the kidney, manifesting as interstitial nephritis and nephrotic syndrome. Bottom line: AKI is common; some estimate that NSAIDs account for about 16% of drug-related AKI.
There is also a strong suspicion (that weathers all sorts of criticisms of the mostly observational data that support it) that CKD incidence and progression to ESKD are potential results of NSAID use, especially when used in context of other illness (a sort of “last-straw” injury that tips people over the edge of tubular resilience).
Now, before we gallop away into the sunset as knephrology knights who have again defended the honor of the nephron from fire-breathing NSAID dragons, we have to hold our proverbial horses. Practicing evidence-based medicine means accepting data that do not always fit with our preconceived or pathophysiologically plausible notions of drug safety. The studies on NSAIDs and CKD are actually extremely mixed; many show no risk or low risk associated only with high NSAID dose over long periods of time. Furthermore, all of the hemodynamic and electrochemical effects of NSAIDs are not applicable to anuric dialysis patients, in whom their toxicity is really limited to GI bleeding.
A quick review of this evidence is also nicely summarized in this recently published brave defense of NSAIDS.
Data from the 1990s suggested that yes, risk of CKD development was higher among NSAID users. In those studies, the risk of ESRD is shown to be 2 to 8 times higher among high lifetime NSAID users compared to nonusers. Then two large cohort studies in the early 2000s, around the time when COX-2 selective inhibitors were getting really hot, showed otherwise, detecting no association. One was an analysis from the Nurses’ Health Study that showed no kidney function decline over 11 years among 1,697 female NSAID users. Their average age was 56 and average GFR at start of study was 88 mL/min/1.732. The other study, done in just over 4,000 “apparently healthy” men, also showed no association between kidney function loss and NSAID use over 14 years of monitoring. Both studies relied on questionnaire / self-reporting, which raises the usual grumblings of recall bias (though it must be said that prescription databases are not perfect data either, given the prevalence of OTC NSAIDs).
After that, Gooch and colleagues performed a study looking at the same question in people a little bit older and with a little bit lower GFR. In that community-based study, just over 10,000 people over 66 years of age with mean GFR 63 mL/min/1.732 were followed for almost three years. There was a linear relationship between cumulative NSAID dose and mean GFR. High-dose NSAID users (defined as greater or equal to ibuprofen 700 mg or naproxen 300 mg daily) were seen to have a 26% increased risk of GFR loss greater than 15 mL/min/1.732 (the primary outcome). Interestingly, NSAID use was not associated with rapid progression among people whose GFR was already < 60 mL/min/1.732 at the time of exposure. This may reflect more cautious use in this group. The authors acknowledge that they used very conservative definitions, including people as “NSAID users” if they had used even very low doses; this may have confounded the apparent trends in the CKD 2+ group, who, you would think, would be most vulnerable to the ischemic vasoconstrictive hit of NSAIDs.
So, a mixed bag, but on the whole our field seems to maintain faith that NSAIDs are generally bad news bears for the kidney set.
We know what you’re thinking. Everyone sorta kinda already knew all that. How ‘bout some NSAIDS 102:
- What about the selective COX inhibitors? Nope, those are bad too, though there is a possibility that they are not AS bad as the nonselectives; for example, in the PRECISION trial, poor renal outcomes in people with high baseline CKD risk (from diabetes, hypertension, and obesity) randomized to ibuprofen 2,400 mg daily were almost twice as high as those randomized to celecoxib 200 mg daily for arthritis pain.

Visual Abstract by @divyaa24 on Nissen et al
- Do certain NSAIDs cause more harm than others? The Most-Wanted list includes phenacetin (the culprit likely responsible for decades of unshakeable NSAID animosity from nephrologists, given its high rates of papillary necrosis that led to high rates of ESRD [it was removed from the market by the FDA in 1983]). Another study showed that ketorolac and oxicams were more likely to cause new CKD than other NSAIDs, with no change in the association due to diabetes mellitus or hypertension. This is interesting, as these agents also have the highest rates of other non-kidney adverse effects, including GI bleeding, and may be related to the duration of action (oxicam has longest half-life of all NSAIDs). Also interesting because of the incredible increases in ketorolac being used in the emergency department as a substitute for opioids. The study suggested a 2.5 times increased risk of CKD even after short-term use of ketorolac.
- Can NSAIDs be used in anuric patients? Actually, this is a rare shiny spot on the NSAID gloomcloud in nephrology. Many of the hemodynamic and electrochemical concerns (decreased perfusion, sodium retention, worsened hypertension and edema, hyperkalemia) are as applicable in anuric patients on dialysis. However, a retrospective study from Taiwan demonstrated an association with strokes in patients on hemodialysis taking NSAIDs. Additionally, we know that the most common pain type among this same group is nociceptive, from musculoskeletal causes, which often (though not always) responds to NSAIDs. Now, with a nod to the myriad nonpharmacological options that are underused for these types of pain, we should embrace the potential for benefit of NSAIDs in this subgroup, especially when given in short courses as discussed in this paper from Koncicki et al. Another plus: NSAIDs may also synergize with opioids, allowing use of lower doses of opioids to gain pain control. Side note: the effect of NSAIDs in dialysis patients with residual renal function has not been studied; for now, we will have to resort to our best judgment (dusty old thing!) and shared decision-making about the risk/benefits of NSAIDs for people who still pee.
- What about topical NSAIDs? Yes, worth a try! There are gels, sprays, and creams that deliver local relief. They work better for acute pain than chronic, but a systematic review of four RCTs did show that topical 1.5% diclofenac was more effective than a knee brace for osteoarthritis. The only fly in the ointment (ha!) is the possibility of systemic absorption, which has been linked to kidney toxicity in case reports like this one. However, these are rare (i.e., case reports) and pharmacokinetic data suggests that only a very modest amount gets absorbed, which likely has minimal effects on kidney blood flow.
So, there you have it. The portrait of a villain, amended and at least partially exonerated by thorough review of its background. For NSAIDs, the fundamental tenet of all pain management holds true – when used thoughtfully, via shared-decision making and in concert with nonpharmacologic techniques, NSAIDs may actually become more friend than foe.

Visual Abstract by @whatsthegfr on Alsaad and Wadei
Check out this podcast episode of The Curbsiders featuring
@Nephro_Sparks@DavidJuurlink@SammyG :#146 NephMadness: Pain Meds in Chronic Kidney Disease
GABAnergics
In treating neuropathy, that lancinating pain that nibbles upward on patients with diabetes and circumferentially during postherpetic neuralgia, we face the very essence of doctoring, summed up by the oft repeated (but somehow unattributed) aphorism: cure sometimes, comfort always. It’s not easy. Knowing the pharmacokinetics, pharmaceutical history, and evidence supporting this class of medications will equip you with the fundamentals needed to resist nihilism.
Let’s go back to the basics: these meds are structural analogs of the neurotransmitter γ-aminobutyric acid (GABA), which ultimately diminishes pain transmission by reducing glutamate-mediated signaling. Their specific mechanism of action is to act as alpha-2-delta ligands that bind the L-type voltage-gated calcium channel blocker in the CNS, reducing glutamate and thereby reducing transmission of nerve pain signals. They are cleared by the kidney and therefore must be dose reduced in those with decreased kidney function. Neurotoxicity (manifesting as somnolence, dizziness, feeling and/or acting like a zombie, myoclonus, and eventually obtundation) are relatively common, especially when used or not appropriately dosed in people with advanced age and multiple comorbidities.

Copyright: CI Photos / Shutterstock
Baclofen is a GABA agonist specific to the GABAB class that − while not intended to be prescribed for pain − is increasingly seen for a variety of indications including hiccups, spasms, and pain syndromes. Since baclofen is excreted by the kidneys, neurotoxicity is common and oftentimes can lead to severe neurological sequelae such as coma. As such, current dosing guidance emphatically recommends against the use of baclofen in patients with ESKD and to reduce dosage based on kidney function.
Our two most widely used GABAnergics for pain syndromes are pregabalin and gabapentin. Pregabalin is FDA-approved for treating diabetic neuropathy (keeping company with two other FDA-approved agents: duloxetine, a selective norepinephrine reuptake inhibitor (SNRI), and tapentadol, which like tramadol is a combined opioid agonist and SNRI). Several randomized controlled trials showed benefit (defined as 50% decrease in pain) of pregabalin over placebo at doses of 300-600 mg daily (among people with normal kidney function).
The most recent position statement from the American Diabetes Association encourages physicians to use pregabalin or duloxetine as first-line for neuropathic pain in diabetes, with gabapentin as an alternative initial approach, basically if the patient can’t afford the others (“taking into account patients’ socioeconomic status, comorbidities, and potential drug interactions”). The CDC guidelines for non-opioid treatment of chronic pain takes a similar stance, recommending gabapentin, pregabalin, and duloxetine as first-line agents for neuropathic pain. However, there’s a problem with their approach. Inexplicably, in the same recommendation, the CDC also refers to these meds as “disease-specific” treatments:
“Use disease-specific treatments when available (e.g. gabapentin/pregabalin/duloxetine for neuropathic pain”
Say what?? Neuropathic pain is not a specific disease – it’s a description, a phenotype downstream of markedly disparate pathophysiologic processes. Take two of the most common life-ruining “neuropathies” – diabetic peripheral neuropathy and lumbar radiculopathy (i.e., “sciatica”). The former is a distal symmetric sensorimotor polyneuropathy that manifests as numbness (from axonal loss) and tingling pain (from axonal damage) derived from large and small myelinated nerves. Sciatica, on the other hand, is a totally different process stemming from mechanical impingement and inflammation in the nerve roots as they exit the spine. It is often a stochastic pain syndrome whose natural history infuriates its victims with sometimes untriggered periods of agony followed by remissions.
So, despite the CDC and others’ well-intentioned lumping of them together under the misery umbrella of “neuropathic pain,” these distinct disease processes are probably the reason that gabanergics are often found to be ineffective. The evidence on pregabalin in diabetic neuropathic pain reduction is top notch; meanwhile, there is moderate- to high-quality evidence that the GABAnergics just don’t work for lumbar radiculopathies, and do cause high rates of side effects. Unfortunately, this doesn’t seem to stop us from prescribing these meds for that condition, thereby throwing several hundred pound sandbags on the “risk” side of the risk-benefit see-saw we are always balancing in pain management.
There is growing concern that GABAnergic prescription rates are increasing. In 2016, it was the 10th most commonly prescribed med, with 64 million prescriptions, up from 39 million in 2012. Cause for this is multifactorial – physicians may be trying to follow guidelines and avoid opioids. There may also be more nefarious forces at play here (hmm, what nefarious force has a history of influencing prescribing patterns in subtle often subconscious ways…you guessed it!). In the 1990s, the pharmaceutical company Parke-Davis (a subsidiary of Pfizer) came under a lot of heat (and paid out over $300 million in lawsuit settlements) for illegal promotion of gabapentin for off-label uses, namely, for pain. It had been approved in 1993 as an adjuvant anticonvulsant and had some persuasive data for efficacy in post-herpetic neuralgia. They used teleconferences, CME programs, and paid speakers to promote the drug, which may be why there is a common belief that the drugs are “worth a try” for any number of off-label indications, including anxiety, hot flashes, nystagmus, migraine, and insomnia.
Now, subsequent studies have generated evidence that they can in fact be effective for some of these conditions. For example, within the CKD and ESKD population, there is decent evidence supporting their use in uremic pruritis (often better than antihistamines!) and restless legs (as efficacious as ropinirole). That said, dosing in patients with CKD and ESKD needs to be careful, as the GABAnergics are all renally cleared. Initial doses should be low (gabapentin 100 mg or pregabalin 25 mg daily in CKD; in hemodialysis patients, three times weekly after dialysis). Dose can be uptitrated over time as needed and tolerated. Extra caution should be exercised among patients on other psychotropic medications, since toxicity can be synergistic; co-administration with opioids, for example, greatly increased drug-related death rates in a population-based retrospective study in Canada.
All in all, the GABAnergics have a time and place in the nephrologist’s world, which is full of neuropathy and complex pain syndromes that may respond to these agents. Start low, go slow, and keep an eye out for toxicity.
Tramadol
This one gets its own category. You may have been taught that it is an opioid, perhaps even a “safer” opioid. You may not have been taught anything about it at all, but have seen it being used in that all too common between-a-rock-and-a-hard-to-control-chronic-pain situation with pretty decent effect.

Copyright: Cranach / Shutterstock
First, let’s review its mechanism of action and metabolism. Just to be clear, it is definitely an opioid, with the same primary physiologic effects – analgesia, miosis, cough suppression, respiratory depression, euphoria, and decreased bowel motility. But, it’s not only an opioid. It’s actually a racemic mixture of two enantiomers. The (+) enantiomer (and its metabolite) act as the opioid with affinity at the mu opioid receptor. The (-) enantiomer acts an SNRI, which modulates pain signal transmission as well as mood, like duloxetine or venlafaxine.
Now, metabolism. Tramadol is metabolized by hepatic cytochrome P450 2D6 enzymes, which are variably expressed in different people. This is key to understanding the incredibly variable effect of tramadol in different patients:
- When you give a dose of tramadol, you actually have no way of knowing how much of the drug will be metabolized to its main opioid metabolite, M1, in each patient. The CYP2D6 enzyme is functionally ABSENT in large numbers of people (“poor metabolizers”), up to 7%-10% of white people and 1-2% of Asian people. Because tramadol itself is a very weak opioid agonist (you need M1 to have clinically detectable analgesia), these poor metabolizers (poor poor metabolizers?) get next to zero opioid analgesia from this med, but are at risk for toxicity as dose is increased waiting for the effect to kick in.
- CYP2D6 activity is also highly influenced by other meds, making patients on tramadol highly vulnerable for drug-drug interactions. Strong inhibitors of the enzyme include meds we use all the time – cinacalcet, paroxetine, fluoxetine, bupropion – as well as ingested substances (not grapefruit – that one’s specific to 3A4, but other herbs like goldenseal). If you multiple the list of drugs that influence 2D6 activity by the average 19 daily medications in patients on hemodialysis, we’re basically asking for drug-drug interactions by prescribing tramadol.
So, how is it that a medication with all the risks of an opioid plus the added complexity of finicky metabolism and SNRI activity become thought of as “safer” than other pain meds? The history of tramadol is rather interesting. It was released in Europe in the 1970s and became FDA-approved in 1995 to treat “moderate to moderately severe pain”. At first, it was not considered a controlled substance due to decades of purportedly uncomplicated use in Europe. However, a proactive surveillance committee was put in place to monitor for abuse and harm; this was rather novel, the first time an independent committee was charged with systematically evaluating safety profile of a drug after FDA approval. During the first two years in circulation in the US, abuse was reported as occurring in 2-3 per 100,000 patients, which subsequently declined to <1 per 100,000 patients thereafter.
The surveillance committee was disbanded by 2004, but subsequent data perpetuated concern about its risks; emergency department visits for adverse effects of the drug increased from 10,000 to 26,000 between 2005 and 2010. Toxicity spanned the gamut from usual signs of opioid intoxication (AMS, respiratory depression) to hypoglycemia to seizure, which are not generally associated with opioids. Withdrawal patterns were also described, which included both the typical opioid withdrawal symptoms as well as an atypical withdrawal syndrome that could happen even after slow down titration of dose (symptoms included hallucinations, paranoia, extreme anxiety, panic, confusion, and numbness/tingling in the extremities). Not only that, these withdrawal symptoms were common, reportedly accounting for 40% of all adverse effects of the drug. By 2014, it was added to schedule IV of the Controlled Substance Act. Most other prescription opioids live in schedule II (the lower the schedule number, the higher the risk of harm or abuse from the drug).
Tramadol is included in the “use with caution” category of the WHO analgesic ladder in CKD. Its unpredictable pharmacokinetics should concern us, as well as its lowering of the seizure threshold (already thought to be lowered in the uremic milieu). Dose adjustment is encouraged. In CKD, an increase in normal dose interval from every 4-6 hours to every 8-12 hours is encouraged, as well as a maximum total daily dose of 200 mg. In dialysis, max dose recommended is 50 mg every 12 hours, dosed after dialysis.
In summary, tramadol is a quirky, shapeshifting opioid-antidepressant combo that produces variable results in different people and doesn’t play well with other (drugs). It should by no means be thought of as a “safer” option than other pain meds. Like all of them, it can be used to decent effect when carefully dosed, monitored, and understood as a pharmacologically and historically complex player in the pain game. Take a look at this Twitter thread on tramadol from our Selection Committee Member, David Juurlink:
“What’s wrong with tramadol?” is something I get asked a lot
Answer: Plenty
Thread ->
— David Juurlink (@DavidJuurlink) October 21, 2017
– Executive Team Member for this region: Matthew Sparks, AJKD Social Media Advisory Board member. Follow him @Nephro_Sparks
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC point for reading this region.
- Register/log in to the NKF’s Professional Education Resource Center (PERC).
- Review the activity and accreditation information.
- Click “Continue” and review Course Instructions.
- Complete Post-Test. Please note: By selecting “Yes” to the participation questions for each region, the corresponding Post-Test questions will appear. Click “Save Draft” to save your responses and finish later. When you are ready to submit your answers, click “Preview” to review all responses, then click “Submit.”
- Click “Next” to complete the Evaluation form, then click“Submit.”
- Claim 1.0 CME credit and 1.0 MOC point per region (up to 8.0 total for 8 regions of NephMadness).
- Print your certificate.
- Review the Post-Test answers and rationale.
The CME and MOC activity will expire on June 15th, 2019.
Submit your picks! | #NephMadness | @NephMadness | #PainRegion
Leave a Reply