
#NephMadness 2020: SGLT2i Region
Submit your picks! | NephMadness 2020 | #NephMadness | #SGLT2iRegion
Selection Committee Member: Meg Jardine @jardine_meg
Meg Jardine is Program Head of Innovation & Kidney Research at The George Institute, Head of Renal Trials at George Clinical, and a practicing nephrologist. Her interests include preventing the progression and complications of kidney disease and diabetes. Dr Jardine was the Global Scientific Lead of the CREDENCE study. She leads the RESOLVE, BEAT-Calci, ACTIVE, and FINESSE trials and has a particular interest in using innovative trial designs to better generate evidence to improve outcomes.
Writer: Harish Seethapathy @BetterCallSeeth
Harish Seethapathy is a nephrology fellow at Massachusetts General Hospital and a 2019-2020 AJKD Intern. His primary research interest is in onconephrology and his work focuses on utilizing epidemiology to study the kidney related toxicities of cancer drugs.
Competitors for the SGLT2i Region
TG Feedback vs Non-TG Feedback
SGLT2i in Transplantation vs SGLT2i Without Diabetes Mellitus

Copyright: Who is Danny / Shutterstock
TG Feedback vs Non-TG Feedback
Stephen King, the prolific American author of horror and supernatural fiction was having a conversation with the not-so-prolific George R.R. Martin, the author of the ‘Game of Thrones’ series about writing speed (King writes 6 pages every day) and fan expectations.
GRRM: How are you so productive and how do you deal with fan pressure?
King: The pressure that you or I feel in getting the next book out there is nothing compared to the pressure on JK Rowling for the 7th Harry Potter book….
In many ways, the pressure was even stronger on the CREDENCE investigators. Angiotensin converting enzyme inhibitors (ACEis)/angiotensin receptor blockers (ARBs) had been game changers since the early 2000s, but for nephrology, a string of failed therapies trying to halt the progression of kidney disease had taken a toll (highlighted in this NephJC summary). It felt like nephrologists were using daggers to fight the fire breathing dragon called diabetic kidney disease (DKD).
While advances had been made in the treatment of various glomerulonephritides, the mantra for DKD seemed to be fixed on the time-tested but archaic principles of managing any kidney disease, namely blood pressure control, proteinuria reduction, and use of ACEI/ARBs. However, this approach has an unsatisfactory high rate of kidney failure and death. Patients in the ARB arm of IDNT had a ~10% risk of death or dialysis in two years; it was ~15% in RENAAL.
The scale of the problem is enormous: 15 million people in the United States (US) have an eGFR <60 mL/min/1.73 m2. Worldwide, the number of patients on dialysis is expected to double, to more than 5 million people, by 2030. An equal number of people will face premature death with a need for dialysis but without access to it. More than half of patients receiving dialysis for end-stage kidney disease (ESKD) have diabetes. The onset of kidney disease in diabetes heralds the onset of cardiovascular deterioration; atherosclerotic and heart failure events increase with deteriorating kidney function.
Finding ways to improve outcomes in DKD has been frustratingly hard for researchers. Lab-grown insulin-producing cells may one day cure type 1 diabetes but, meanwhile, nephrologists are tasked with slowing the progression of kidney disease while minimizing the risk of cardiovascular (CV) complications. Surprising many, the cardiovascular safety trials showed that sodium glucose co-transporter-2 inhibitors (SGLT2i) agents may fit the bill. CANVAS and EMPA-REG not only showed CV protection but hinted at powerful reduction in kidney endpoints. The stage was set for CREDENCE, and it did not disappoint, with data showing profound nephroprotection. Nephrology will never be the same again.
And so, to compete spectacularly in what has to be the most unsurprising NephMadness region of 2020, we have mechanisms and new indications of SGLT2 inhibitors.
Tubuloglomerular Feedback (TGF)
The best way to understand the nephroprotective mechanism behind SGLT2 inhibitors is to look at the last kidney savior, RAAS blockade. Hyperglycemia is well known to promote glomerular hyperfiltration in DKD. Lowering caloric intake or weight loss after bariatric surgery has been shown to reverse this aberrance in kidney hemodynamics. While the pathogenesis of hyperfiltration is incompletely understood, it is hypothesized that activation of the RAAS leading to afferent arteriolar vasodilation and efferent arteriolar vasoconstriction increases intraglomerular pressure and this sends the kidney down the road of irreversible damage.

Copyright: Zuzana Semanova / Shutterstock
To date, RAAS blockade has been the last successful drug class for GFR preservation, independent of the effect on blood pressure. That success takes us back almost two decades, to 2001, when the RENAAL and IDNT trials showed that losartan and irbesartan, respectively, reduced the composite kidney endpoint (ESKD, doubling of serum creatinine, and mortality) by 15-20%. Unfortunately, diabetic kidneys subject to RAAS blockade still progressed to kidney failure at a fairly significant pace.
“Man had acquired a past; and he was beginning to grope towards a future.” – Arthur C. Clarke, 2001: A Space Odyssey
After years of trying, we discovered (stumbled upon) an unlikely hero, the SGLT2 inhibitors. Sodium glucose transporters are located in many tissues, and SGLT1 and SGLT2 play a central role in kidney glucose homeostasis. Glucose is freely filtered at the glomerulus, and then the majority of the filtered glucose (~80% of the 180 g/day) is reabsorbed through the low-affinity, high-capacity, SGLT2 co-transporter (1:1 Na:glucose absorption) in the early proximal convoluted tubule while the high-affinity low-capacity SGLT1 co-transporter reabsorbs the final 10-20% in the late proximal tubule.
In patients with diabetes, SGLT co-transporters are upregulated 3-4 fold, resulting in increased kidney glucose resorptive capacity. This made these transporters attractive therapeutic targets to improve glycemic control. Selective SGLT2 inhibition was preferred as additional SGLT1 inhibition with the natural compound phlorizin led to increased gastrointestinal side effects. Phlorizin, interestingly, is isolated from apples (as the saying goes…”An apple a day keeps the doctor away”). Maximal SGLT2 blockade only blocks around 30-50% of the filtered glucose load (~80 g/day or 320 kcal/day), due largely to upregulation of SGLT1 mediated glucose resorption in the late proximal tubule.
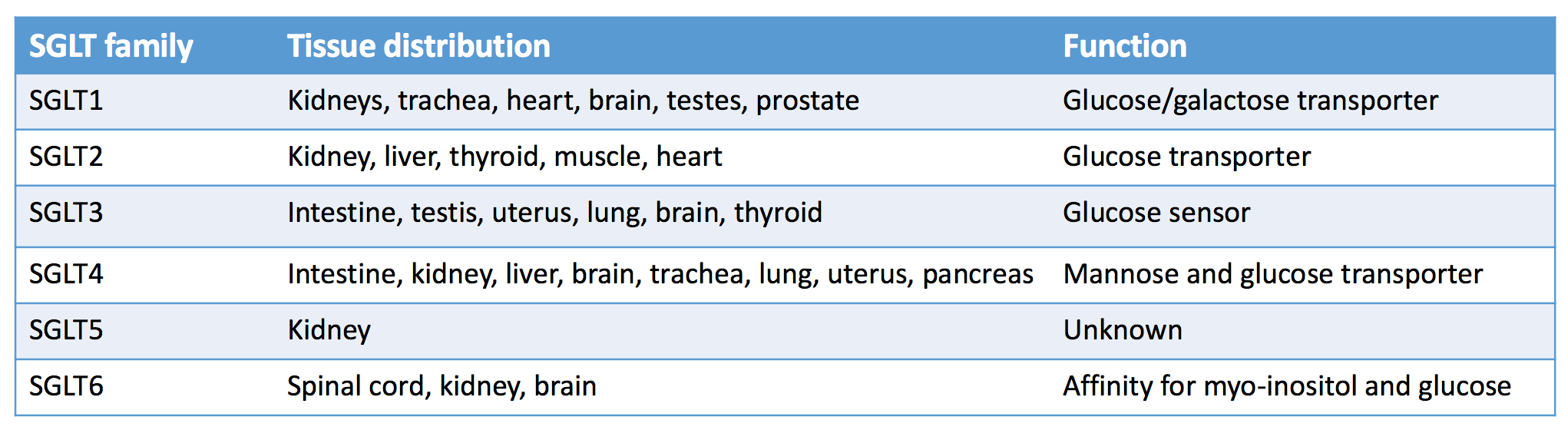
Distribution of sodium glucose co-transporters in the human body. Adapted Table 2 from Kalra et al, © Indian J Endocrinol Metab.

SGLT receptors in the kidneys. Figure 1 from Alicic et al, AJKD © National Kidney Foundation.
When the first clinical outcome trials on SGLT2 inhibitors (CANVAS, EMPA-REG , and DECLARE-TIMI) were published, it quickly became obvious that SGLT2 inhibitors were not merely glucose-lowering drugs. Yes, there were small decreases in HbA1C (~0.5%), blood pressure (~2-5 mm Hg), and weight (~2-3 kg), but these modest effects could not fully explain the reduction in CV events and mortality. Though kidney endpoints were not the primary outcomes in the early trials, hints of nephroprotection were seen in secondary endpoints and post-hoc analyses. A kidney-centric trial (ie, CREDENCE) was of utmost importance as there were doubts if the drug would work at lower eGFR levels (<45 mL/min). CREDENCE confirmed the benefit of canagliflozin in patients with proteinuria and eGFR down to 30 mL/min (curiously, a level at which its effect on glucosuria is minimal).

Major SGLT2i trials before CREDENCE
It is important to remember that the majority of patients (~80%) in the early SGLT2i trials were on an ACEi or ARB and all of the patients in CREDENCE were on maximally tolerated doses of ACEi/ARB therapy. So, SGLT2 inhibitors work in patients with DKD on RAAS blockade, which produces efferent vasoconstriction, but the crucial question remains: Why?
The most commonly postulated mechanism, and one that was first demonstrated in animal models using phlorizin, involves tubuloglomerular feedback (TGF). The macula densa with the juxtaglomerular apparatus (JGA) maintains a stable glomerular filtration rate by sensing distal sodium and chloride delivery and modulating changes in the afferent arteriolar tone. In DKD, a multistep process results in the kidney misunderstanding an individual’s volume status and consequently, in hyperfiltration:
- Persistent hyperglycemia upregulates glucose reabsorption (and secondarily, sodium and chloride reabsorption) in the proximal tubule
- This decreases sodium and chloride delivery to the macula densa which is mistaken for decreased kidney perfusion
- This results in decreased adenosine production
- A decrease in afferent arteriolar tone increasing blood flow to the glomerulus is signaled
When patients start an SGLT2 inhibitor, the process is reversed:
- Glucose (as well as sodium and chloride) reabsorption is blocked in the proximal tubule
- This increases sodium delivery to the thick ascending limb of the loop of Henle
- This increases the activity of the Na-K-2Cl co-transporter, an energy-requiring process where adenosine triphosphate (ATP) is broken down to adenosine
- Increased adenosine acts in a paracrine manner on the afferent arteriolar smooth muscle cells and leads to afferent arteriolar vasoconstriction, decreasing blood flow to the glomerulus
- This afferent vasoconstriction combines synergistically with the efferent vasodilation produced by RAAS blockade to reduce intraglomerular pressure and glomerular filtration
This phenomenon can be recognized in the first few weeks of SGLT2 inhibitor treatment where there is an acute reduction in GFR followed by stabilization. This acute drop in GFR can be seen after a single dose of SGLT2i showcasing the acute nature of tubuloglomerular feedback. Trial data shows that this effect is completely reversible after a 30-day washout period.

Change in eGFR from baseline. Adapted Figure 3B from Perkovic et al, NEJM © Massachusetts Medical Society.
Non-Tubuloglomerular Feedback
Occam’s razor: No more things should be presumed to exist than are absolutely necessary; i.e. the fewer assumptions an explanation of a phenomenon depends on, the better the explanation.
Critics decry the attribution of the tremendous benefit of SGLT2i to TGF alone. If TGF was the only play at hand, NSAIDs which also lead to afferent vasoconstriction would be our dear friend. And how can the consistent benefit on CV outcomes across the spectrum of eGFRs be explained? Shouldn’t SGLT2 inhibitors be ineffective in patients with lower eGFRs with reduced tubular drug concentrations? Thankfully, we have critical thinkers in medicine who ignore Occam’s razor and instead abide by Hickam’s dictum.

Copyright: Sergey Nivens / Shutterstock
We already know that SGLT2 inhibitors produce caloric losses that translate to weight loss and improved A1C. The average weight loss with these drugs is around 2-3% of the body weight. SGLT2 inhibitors also lead to chronic osmotic diuresis due to loss of glucose and water, which lead to fluid losses (this glucosuria puts patients at a higher risk for genital mycotic infections). This is demonstrated by the increase in urine output from baseline after SGLT2i initiation. This diuresis may also help blood pressure control, with all SGLT2i trials showing a consistent 2-5 mm Hg lowering of blood pressure compared to controls. Besides these obviously beneficial non-TGF pathways, there are other hypothesized mechanisms for the benefits of SGLT2i. Let’s take a look at a handful.
Reduction of inflammation and fibrosis:
Increased intracellular glucose concentrations in the proximal tubules of patients with diabetes mellitus (DM) induces inflammatory cytokines, fibrotic mediators, and reactive oxygen species. This was demonstrated nicely by exposure of a human proximal tubular cell line to empagliflozin showing reduction of inflammatory and fibrotic signals (TLR4, NF-κB, IL-6 and collagen IV); this may potentially play a role in ameliorating nephron loss caused by inflammation and fibrosis. Another recently recognized pathway involves kidney VEGF-A expression. In a kidney ischemia-reperfusion injury mice model, SGLT2i protected against kidney ischemia and prevented interstitial fibrosis by upregulating VEGF-A expression.
Alteration of oxygen consumption and lowering cortical hypoxia:
The kidneys are second only to the heart when it comes to oxygen consumption. This is almost entirely due to the high oxygen requirement of the Na-K-ATPase. In diabetes, this energy consumption is enhanced by SGLT2 overactivation, proximal tubular hypertrophy, and hyperfiltration. Animal models using phlorizin have shown that SGLT2 inhibition suppresses the activity of Na-K-ATPase and reduces total oxygen consumption by decreasing intracellular glucose, sodium concentration, and proximal tubular hypertrophy. There is a catch, though: reducing sodium reabsorption in the early proximal tubule decreases oxygen consumption in that segment, but increases it in the thick ascending limb, collecting ducts and medulla.
The consequences of this increased distal oxygen consumption are not entirely clear. It was thought that reduction in cortical hypoxia would help recover erythropoietin producing fibroblasts and lead to an increase in hematocrit, further reducing hypoxia. But this hypothesis could not be confirmed in an animal model. While the hematocrit rise seen with SGLT2i can be partially explained by hemoconcentration from osmotic diuresis, the full picture remains unclear.
The redistribution of hypoxia in the kidneys also affects HIF-1α expression in proximal tubular cells. Stable HIF-1α expression leads to interstitial fibrosis. SGLT2 inhibitors have been shown to inhibit expression of HIF-1α and its target genes (such as VEGF, GLUT1, PKM, etc) while promoting its degradation by suppressing mitochondrial oxygen consumption. This prevents HIF-1α induced interstitial fibrosis in the diabetic kidneys.
Alteration of energy metabolism:
Energy utilization is impaired in diabetes and there is an over-reliance on fatty acids for energy production. This is inefficient. SGLT2 inhibitors alter the dynamics of energy metabolism by inducing a fuel shift in the kidney tubular cells from free fatty acids to hydroxybutyrate, considered a ‘superfuel’ as it produces more energy per unit oxygen consumption. This is also important in the heart. Utilization of ketones prevents the accumulation of fatty acid intermediates that promote lipotoxicity and diastolic dysfunction.
Cardioprotective effect (secondary renoprotection):
In addition to decreased ventricular loading due to a decrease in preload, SGLT2 inhibitors also have direct effects on myocardial tissue through the following mechanisms, mostly studied in heart failure.
-
- SGLT2 inhibitors also inhibit sodium hydrogen exchangers. Heart failure models show that SGLT2i block NHE1 in the myocardium (despite no SGLT2 receptors in myocytes) and NHE3 in the proximal tubule. NHE1 inhibition reduces intracellular sodium and calcium which decreases pro-oxidant and pro-thrombotic states. NHE3 inhibition promotes natriuresis.
- Conversion from fatty acids to beta-hydroxybutyrate improves cardiac energy efficiency as mentioned above.
- Anti-fibrotic effects by suppressing collagen synthesis, inhibiting myofibroblast differentiation and reducing pro-inflammatory cytokines (IL6). This directly translates to reduced LV mass and improved ejection fraction.
SGLT2i-induced inhibition of the sympathetic nervous system has also been purported as a potential mechanism by which they improve CV outcomes. With a continual output of new literature about possible mechanisms coming our way, we will learn more in the future.

Mechanisms other than TGF

Effect of SGLT2 inhibition on different organs. Adapted Figure 1 from Wanner et al, Diabetologia © Springer-Verlag GmbH Germany.
So it should be clear that SGLT2 inhibitors exert their kidney benefits through diverse mechanisms whose individual effects are hard to quantify and their overall benefit involves interplay between different organs. Considering we still don’t know how some of the most common drugs work, as well as the monumental difficulty associated with new drug discovery, perhaps we should be grateful that SGLT2 inhibitors work and just enjoy debating the smaller problem of figuring out how they work.
Check out this podcast episode of The Curbsiders featuring @BetterCallSeeth, @Nephro_Sparks, and @kidney_boy:
#204 SGLT2 Inhibitors
SGLT2i in Transplantation vs SGLT2i Without Diabetes Mellitus
With SGLT2 inhibitors now expected to be used widely in patients with diabetes and CKD, the obvious next question is: Can SGLT2i be used with the same degree of success for other patient populations such as those who are post-kidney transplant or who have non-diabetic CKD?

Trade card for Hunt’s Remedy, the great kidney & liver medicine, William E. Clarke, proprietor, Providence, Rhode Island, undated. Courtesy of Historic New England.
SGLT2 Inhibitors in Transplantation
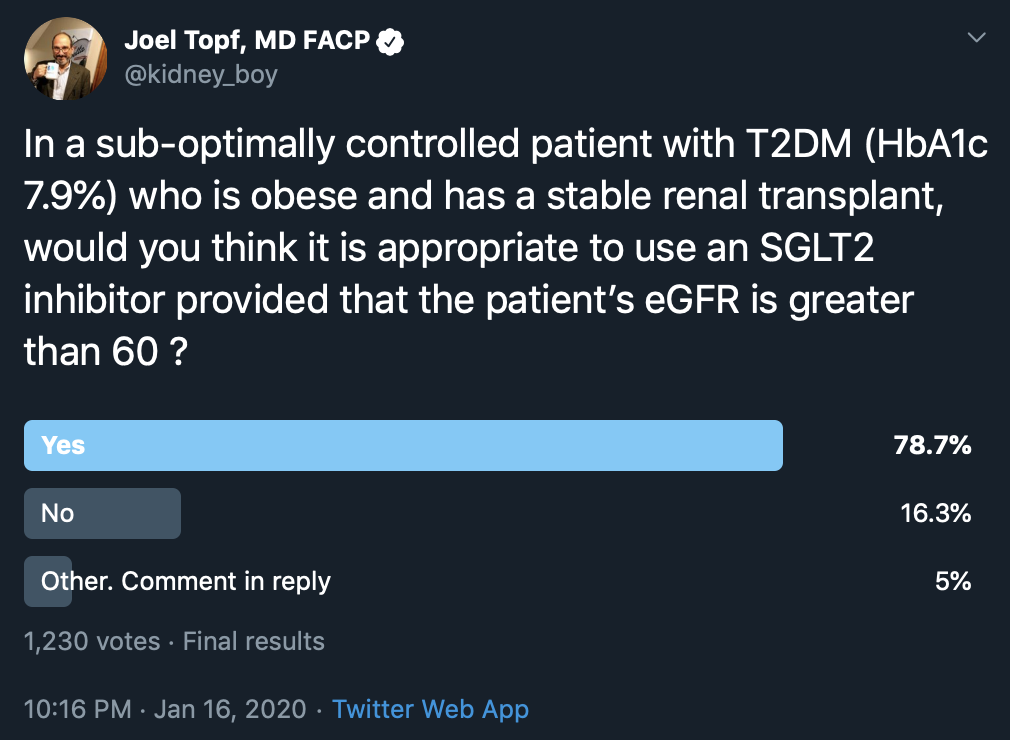
Post-transplantation DM is fairly common (an incidence of 10-20%) and is associated with adverse graft outcomes and increased CV mortality. Can SGLT2 inhibitors be used in this condition? In order to gain more insight, Beshyah et al (in the pre-CREDENCE era) used a physician response survey to ask the following question:
“In a sub-optimally controlled patient with T2DM (with HbA1C 7.9%) who is obese and has a stable kidney transplant, would you think (on basis of efficacy and safety) it appropriate to use an SGLT2 inhibitor provided that the patient’s eGFR is greater than 60 (i.e., serum creatinine is normal)?”
Out of 44 respondents (primarily from the Middle East), nearly 60% said they use SGLT2 inhibitors in this patient population regularly, while 20% said they would never use it in a transplant patient even if the eGFR were >60 mL/min/1.73 m2. Respondents unlikely to prescribe it were worried about the risk of urinary tract infections (UTIs) in immunocompromised patients, the risk of dehydration, and its impact on patient survival as well as the initial reduction of eGFR with these drugs. The respondents who supported using SGLT2 inhibitors in transplant felt that such risks were small and worth taking considering the CV benefits.
Copyright: Robsonphoto / Shutterstock
A similar question on Twitter generated 1,230 responses
Do we have data to support the use of SGLT2 inhibitors in transplant patients? First, a reminder that the major SGLT2i trials did not enroll any transplant patients. There have been smaller studies in transplant patients – two clinical trials and two retrospective case series. The prospective studies reported the efficacy of empagliflozin in transplant patients with post-transplant DM: the Vienna study, a single-arm trial (completed n=14) and the Oslo study, a double-blinded RCT (study arm n=22). The retrospective case series (Rajasekeran et al and Mahling et al) looked at the use of canagliflozin post-transplant.

Studies of SGLT2 inhibitors in transplant patients

Visual Abstract by @Vernisartan on Halden et al
In the limited evidence we have available, SGLT2 inhibitors appear to be safe with no major effect on graft function or levels of immunosuppressive medications. While the risk of UTI does not seem to be increased, it needs to be evaluated in larger studies before definite conclusions can be made.
There are no prospective studies looking at cardiovascular or other hard outcomes (i.e. mortality) in transplant patients with diabetes or kidney outcomes such as progression to ESKD in patients who develop chronic allograft nephropathy after transplantation. With the current American Diabetes Association (ADA) recommendation to use these agents in patients with type 2 diabetes and cardiovascular or kidney disease, it is likely that we will see clinicians extrapolate the benefit derived from SGLT2 inhibitors in non-transplant patients and extend their use to solid organ transplant patients in the coming years. Post-transplant DM is more common after other solid organ transplants (20-30% after heart transplant, 20-40% after lung and liver transplant) so this question will play itself out in all types of transplants. While future data may support this use as appropriate, clinicians would be wise to learn from the cautionary tales of other drugs that were proven to be beneficial in non-transplant kidney disease but were shown to lack benefit (ramipril) or be of less benefit than expected (statins) in transplant patients.
SGLT2i Without Diabetes Mellitus
Ah, here comes a twist. One’s pick from the previous matchup on the mechanisms of SGLT2 inhibitors may influence their selections in this matchup. If one believes that SGLT2 inhibitors have a glucosuria-independent mechanism (ie, effects other than tubuloglomerular feedback), then they may be biased towards this candidate in our matchup – the use of “SGLT2i in non-diabetic CKD” is entirely based on the premise that non-glycemic pathways are important.
“Cutting the cord with diabetes”
While it makes physiological sense that SGLT2 inhibitors should be used to manage DKD, the diverse mechanistic pathways and plethora of beneficial effects make us wonder how relevant diabetes is to the discussion. Remember the animal models of SGLT2i showing an increase in VEGF-A leading to protection against kidney ischemia? Those studies were done in non-diabetic rats. Another important study looking at the influence of dapagliflozin was also demonstrated in a non-diabetic mouse model. Two findings stood out in that study:
- SGLT2 is expressed by podocytes and the expression is upregulated in proteinuria (in isolation). This was confirmed in human podocytes and in biopsy samples of patients with CKD and proteinuria.
- Dapagliflozin was shown to limit podocyte dysfunction and loss through reorganization of the actin cytoskeleton of the podocytes.
This effect of SGLT2i on preservation of podocyte structure may have important implications. Perhaps, you don’t need to have diabetes and an upregulation of proximal tubular expression of SGLT2 for these drugs to have an impact. Taking this into account, the makers of the drugs have wasted no time in initiating clinical trials for other indications regardless of diabetes status. EMPA-KIDNEY and DAPA-CKD are now underway to test the efficacy of empagliflozin and dapagliflozin in reducing kidney endpoints, enrolling both patients with and without diabetes.

Copyright: Brian A Jackson / Shutterstock
Multiple trials also are underway to test the efficacy of these drugs in heart failure as well, with diabetes status not a criteria for enrollment. One such trial, DAPA-HF, was recently completed: dapagliflozin was studied in patients with reduced ejection fracture and more than half the patients (55%) did not have diabetes. It continued the trend of significant CV benefit with an 18% reduction in CV death in the treatment group and the benefit was similar in patients with and without diabetes. While it showed a 29% reduction in the kidney composite outcome, the result was not statistically significant, likely due to the small number of events and short trial duration.
The use of these agents in patients without diabetes is likely to be a hot discussion in the coming years. With the benefits of SGLT2 inhibitors well-established in patients with diabetes, enrolling them in ongoing/future trials will be difficult, given the lack of equipoise. Hence, patients without diabetes are likely to make up a large proportion of the patients enrolled in the ongoing and future trials.

Ongoing SGLT2 inhibitor trials with kidney outcomes
– Executive Team Member for this region: Joel Topf, AJKD Social Media Advisory Board member. Follow him @kidney_boy.
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC point for reading this region.
- Register/log in to the NKF’s Professional Education Resource Center (PERC). If you select “Physician” in the drop-down menu during registration, the ABIM ID will pop up – make sure to complete this during registration to receive MOC points after course completion.
- Review the activity, disclosure, and accreditation information.
- Click “Continue” and review Course Instructions.
- Complete Post-Test. Please note: By selecting “Yes” to the participation questions for each region, the corresponding Post-Test questions will appear. Click “Save Draft” to save your responses and finish later. When you are ready to submit your answers, click “Preview” to review all responses, then click “Submit.”
- Click “Next” to complete the Evaluation form, then click“Submit.”
- Claim 1.0 CME credit and 1.0 MOC point per region (up to 8.0 total for 8 regions of NephMadness).
- Save/print your certificate.
The CME and MOC activity will expire on June 13th, 2020.
Excellent Review of role of SGLT2 inhibitors from a Nephrology Perspective