
Test Your Knowledge: HIV and The Kidney
In a quiz published in AJKD, Lum et al (freely available) describe a case of acute kidney injury in a patient with well-controlled HIV infection on highly active anti-retroviral therapy (HAART). The following questions will test your pathology knowledge on HIV-associated kidney disease.
Post prepared by and all images courtesy of Tibor Nadasdy, MD, AJKD Blog Contributor and AJKD Kidney Biopsy Teaching Case Advisory Board member.
For a PDF version of the questions & answers, please click here.


1. A 39-year-old African American woman with CKD stage 3 and sickle cell disease presented to the emergency room with nausea, vomiting, headache, and abdominal pain. Her systolic blood pressure was 165/100. Serum creatinine was 2.2 mg/dL with an eGFR of 26 mL/min. Hemoglobin was 8 g/dL, platelet count 220,000, LDH elevated at 560, and haptoglobin levels were undetectable. Urine protein/creatinine ratio was 6 g/mg. Urinalysis showed 0-3 RBCs/HPF. Tests for hepatitis B and C were negative. Her HIV test came back positive. ANA was negative. Serum complement levels were normal (C3 112, C4 38). A kidney biopsy was performed. What is the most likely diagnosis?
Severe microvascular changes secondary to sickle cell crisis
Incorrect.
HIV-associated nephropathy (HIVAN)
Incorrect.
Obliterative microvascular changes secondary to malignant hypertension
Incorrect.
Thrombotic microangiopathy (TMA), probably associated with HIV infection
Correct! Panel A (Hematoxylin and Eosin [H&E]) shows glomerular capillaries occluded by amorphous material and fragmented red blood cells. Panel B (H&E) shows a small interlobular artery occluded by amorphous material and fragmented red blood cells. These changes are diagnostic of TMA. Based on the clinical presentation and the morphology, the definitive etiology of the TMA in this patient is difficult to determine; however, TMA is known to be associated with HIV infection, and this patient was recently diagnosed to be HIV positive. TMA is a feared complication of HIV infection and usually causes progressive kidney failure. This patient also developed rapidly progressive kidney failure and shortly after biopsy needed dialysis. The pathogenesis of HIV infection-associated TMA is unclear; some cases resemble atypical hemolytic syndrome, while others are more reminiscent of thrombotic thrombocytopenic purpura with ADAMTS-13 deficiency. Considering that HIV-positive patients commonly develop autoantibodies, it is possible that the TMA in HIV-infected patients is secondary to autoantibody-mediated microvascular damage.
Although this patient has sickle cell disease, the morphologic findings are not that of a sickle cell crisis. In sickle cell disease the small arteries/arterioles and the glomerular capillaries are frequently engorged by sickling red blood cells, but fibrin thrombi and fibrinoid necrosis of the microvasculature are uncommon and fragmented red blood cells are usually not seen. The glomerular changes were not that of HIVAN; there was no evidence of collapsing glomerulopathy. Heavy proteinuria can sometimes be seen in patients with TMA if there is severe glomerular capillary damage. Although changes of TMA may develop in patients with malignant hypertension, this is less likely in this woman as she did not have malignant-level hypertension on presentation. Also, in TMA secondary to malignant hypertension arterial/arteriolar changes of TMA dominate, and glomerular changes of TMA are less common; the glomeruli usually show only signs of ischemia.



2. A 30-year-old African American woman with a history of HIV infection. She has been inconsistent with her HAART therapy. She presented with worsening blood pressure to 152/100, and was found to have a serum creatinine elevated at 3.5 mg/dL (eGFR 19 mL/min). Her urine protein/creatinine ratio was 7.8, and microscopic hematuria was present. Her ANA was positive at 1:320; however, her dsDNA was negative. Her CD4 count was low (40) and her HIV viral load was over 1,000,000. Serum complement levels were normal. Ultrasound revealed normal-sized kidneys. A kidney biopsy was performed. What is the most likely diagnosis?
HIVAN (collapsing glomerulopathy)
Incorrect.
Proliferative lupus nephritis with focal crescent formation
Incorrect.
HIV-associated immune complex glomerulonephritis with lupus-like features
Correct! Panel A (Periodic acid-Schiff [PAS] stain) shows a hypercellular glomerulus. The hypercellularity is both extracapillary (crescent) and intracapillary. Approximately 50% of the nonsclerotic glomeruli had cellular crescents; the remaining glomeruli showed only intracapillary hypercellularity. One-third of the glomeruli were sclerotic, and one-third of the interstitium was fibrotic with some inflammatory cell infiltrate. Immunofluorescence showed mesangial and glomerular capillary deposits with a full-house pattern (IgG, IgA, IgM, C1q [See panel B], C3, and kappa and lambda light chains). Extraglomerular immune complex deposits were not seen. Panel C shows mesangial electron-dense deposits. Scattered subendothelial and intramembranous deposits were seen along the glomerular capillaries. Endothelial tubuloreticular inclusions were easily identified.
Although the patient is ANA positive and the glomerular morphology, including the immune complex deposition with a full-house pattern, is suggestive of proliferative lupus nephritis, the high HIV viral load, the low CD4 count, the normal serum complement levels, and the absence of extraglomerular immune complex deposits is more consistent with HIV-associated immune complex glomerulonephritis. HIV patients can develop glomerulonephritis resembling lupus nephritis, particularly if they have high viral load. In our experience, successful treatment of HIV infection with HAART may result in remission of the glomerulonephritis. The presence of endothelial tubuloreticular inclusions seen by electron microscopy is not helpful in differentiating these diagnosis because such inclusions can be seen both in HIV infection and systemic lupus erythematosus.
The cellular crescents resemble large glomerular epithelial cells filling Bowman’s space in collapsing glomerulopathy (pseudo-crescent of collapsing glomerulopathy), but the entire histology is not that of HIVAN. In HIVAN, immune complex deposits are usually absent and true crescents are not seen. Also, characteristic tubulointerstitial lesions of HIVAN, including microcystic dilatation of the tubules, were not seen either. Pauci-immune (usually ANCA-associated) crescentic glomerulonephritis does not have this degree of glomerular immune complex deposits.
Pauci-immune crescentic and necrotizing glomerulonephritis
Incorrect.
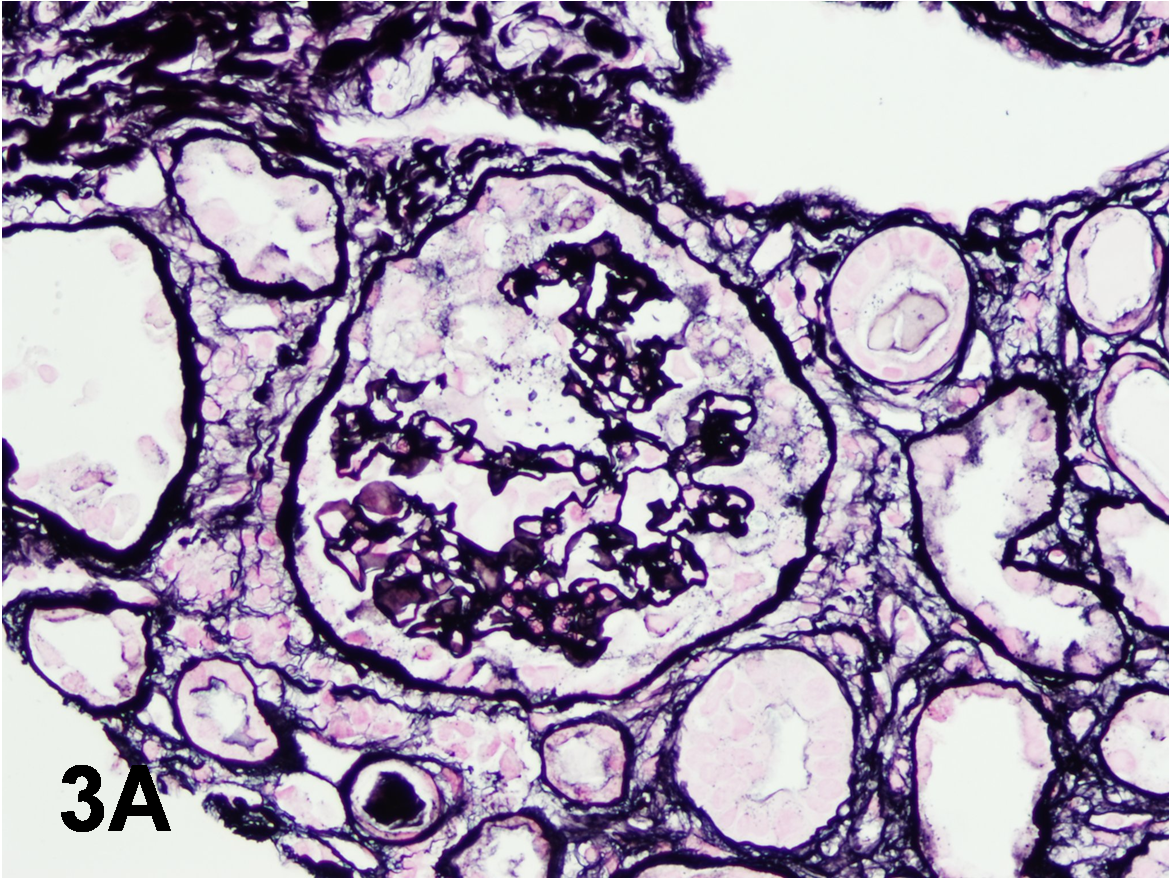


3. A 27-year-old African American man was diagnosed with HIV infection one year earlier and started on HAART therapy. However, he was nonadherent with his medications and presented with a CD4 count of 33 and an HIV viral load of over 9,000,000 copies/mL. His serum creatinine increased to 2.7 mg/dL (eGFR 36 mL/min) from 1.0 mg/dL (eGFR 119 mL/min) over the last month. Urinalysis showed over 300 mg/dL protein and mild microscopic hematuria with 3-4 RBCs/HPF. Twenty-four hour urine collection identified 10g of proteinuria. Serum complement levels were normal. He was negative for hepatitis B and C and syphilis. A kidney biopsy was performed. What is the most likely diagnosis?
Crescentic glomerulonephritis
Incorrect.
HIVAN
Correct! This patient has HIVAN. The kidney biopsy shows typical changes of collapsing glomerulopathy. The glomeruli demonstrate sclerosing/collapsing capillaries with prominent epithelial cells filling Bowman space (Panel A, Jones methenamine silver). In HIVAN, characteristic tubulointerstitial changes, including microcystic dilatation of tubules with a scalloping outline (see Panel B, H&E), are also typically seen. Microcystic dilatation of tubules may also occur in patients with advanced-stage chronic kidney disease of any etiology. However, this patient had normal kidney function just a month before the kidney biopsy; therefore, advanced chronic tubulointerstitial disease is unlikely. Immunofluorescence did not reveal immune complex deposits. Electron microscopy showed several endothelial tubuloreticular inclusions (Panel C). Such endothelial tubuloreticular inclusions are common in patients with HIV infection with or without associated kidney disease.
The hypertrophic glomerular epithelial cells resemble cellular crescents (pseudocrescents); however, in true cellular crescents there is usually glomerular capillary rupture as well as Bowman capsular rupture, which are not seen in collapsing glomerulopathy. Also, fibrin/fibrinogen deposition is usually striking in cellular crescents but not seen in collapsing glomerulopathy. In idiopathic FSGS, collapsing glomerular lesions involving several glomeruli are not seen. Furthermore, in early stages of FSGS the tubulointerstitial disease is mild, microcystic dilatation of tubules is absent, and patients rarely develop acute kidney injury.
HAART treatment was reinstituted, and the patient’s viral load became undetectable within two months. His CD4 count also normalized. Interestingly, his kidney function improved and his serum creatinine stabilized around 1.5 mg/dL (eGFR 73 mL/min). His proteinuria decreased to around 5g/24 h within two months, and later urine protein/creatinine ratio has been between 2.5 and 3. Although HIVAN is usually a relentlessly progressive kidney disease, the example of this patient shows that with optimal and successful treatment of the underlying HIV infection, dialysis can be avoided or at least delayed.
Advanced chronic tubulointerstitial disease secondary to nephrotoxic medications
Incorrect.
Idiopathic focal segmental glomerular sclerosis (FSGS)
Incorrect.


A 52-year-old white man has a history of well-controlled HIV infection, coronary artery disease, diet-controlled diabetes, treated hypertension, and asthma. His medications include lopinovir/ritonovir, emtricitabine/tenofovir, tesamorelin, fenofibrate, olmesartan, albuterol, and promethazine. Recently, he received azithromycin for an upper respiratory tract infection. His serum creatinine gradually rose from 1.0 to 1.1 mg/dL (eGFR of 77-86 mL/min) to 1.5 mg/dL (eGFR of 53 mL/min) over the past year. He presented to the emergency room with nausea, vomiting, and abdominal pain, and was found to have a serum creatinine of 5 mg/dL (eGFR of 12 mL/min). His CD4 count was over 600, and his HIV viral load was undetectable. Serum complement levels were normal. Hepatitis B and C serologies were negative. ANA was negative. Urinalysis showed mild proteinuria, a few RBCs, and numerous WBCs. A kidney biopsy was performed. What is the most likely diagnosis?
Acute myocardial infarction with secondary ATN
Incorrect.
HIV-associated nephropathy
Incorrect.
Azithromycin-induced acute interstitial nephritis
Incorrect.
Acute tubular necrosis (ATN), in the background of interstitial fibrosis and tubular atrophy from a combination of nephrotoxic medications (such as tenofovir)
Correct! It is now well recognized that tenofovir can be nephrotoxic and may lead to ATN. It is debated whether tenofovir can induce chronic tubulointerstitial injury as well. It is intriguing that patients may take tenofovir for some time before they develop acute kidney injury and overt signs of nephrotoxicity. Tenofovir is thought to cause mitochondrial damage in renal tubules. It is likely that factors other than tenofovir exacerbate the acute kidney injury. This patient had an upper respiratory tract infection, was taking over-the-counter medications and antibiotics, and presented with volume depletion. He had a slightly elevated serum amylase and the possibility of acute pancreatitis was entertained, but his serum amylase quickly normalized after volume resuscitation. The kidney biopsy showed clear evidence of ATN with tubular epithelial irregularities and flattening associated with interstitial edema, but also interstitial fibrosis and tubular atrophy (Panel A, H&E). The glomeruli were unremarkable. The interstitial inflammation was mild relative to the tubular injury. Immunofluorescence and electron microscopy did not show immune complex deposits. The glomerular ultrastructure was unremarkable. Proximal tubules, however, lost their brush border, and had abnormally shaped, swollen mitochondria with a reduced numbers of irregularly oriented cristae and flocculent densities in the matrix (Panel B). Although there were some dilated tubules with proteinaceous casts, neither the glomerular nor the tubulointerstitial morphology was consistent with HIVAN. The patient did not have nephrotic syndrome, did not have collapsing glomerular changes, and the tubular dilatation was not the microcystic type with scalloping outline. The interstitial inflammation was mild and patchy with no eosinophil granulocytes; this makes drug-induced acute interstitial nephritis unlikely. The patient did not have cardiac dysfunction, and after volume resuscitation, withdrawal of tenofovir and azithromycin, and three weeks of dialysis, his kidney function started to improve. His serum creatinine returned to 2 mg/dL (eGFR 37 mL/min).
nice questions