
#NephMadness 2023: Thrombotic Microangiopathy Region
Submit your picks! | NephMadness 2023 | #NephMadness | #TMAregion
Selection Committee Member: Anuja Java @anuja_java
Anuja Java is a transplant nephrologist at Washington University in St. Louis, MO. Her research involves functional characterization of genetic variants in complement-mediated diseases. She co-chairs the ClinGen complement gene curation expert panel, is a councilor in Women in Nephrology, has served on the ASN career advancement committee, and received the 2022 NKF “Award of Excellence”.
Writer: Viralkumar Amrutiya @viral_nephro
Viralkumar Amrutiya is a second-year nephrology fellow at Virginia Commonwealth University, Richmond, VA. He is a graduate of Hebei Medical University, Shijiazhuang, Hebei, China, and completed his residency and chief year at Hackensack Meridian Health – Palisades Medical Center in North Bergen, NJ. He has special clinical interest in management of CKD and transplant nephrology. He would like to work towards minimizing disparities and bringing equality in kidney care.
Writer: Nouman Akbar @AkbarNouman
Nouman Akbar is a second-year nephrology fellow at Virginia Commonwealth University. He is a graduate of Providence Hospital, Washington, DC, where he completed his residency and chief year. His clinical interests include lupus, C3GN, TMA, and transplant nephrology.
Competitors for the
Thrombotic Microangiopathy (TMA) Region
Team 1: Primary TMA versus Team 2: Secondary TMA

Introduction
The Thrombotic Microangiopathy (TMA) region encompasses disorders with microvascular endothelial injury associated with thrombotic, hemolytic, and ischemic complications. TMA presents a number of clinical challenges on the court such as thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and organ damage including AKI.
These teams’ lineups are based on the evolving classification of TMA. Here we use Primary TMA (complement-mediated, replacing the older term Atypical HUS) and Secondary TMA (triggered by another disease), as suggested by our region’s expert Dr. Anuja Java in a recent book (Fig 1). Interestingly, there is a long history of trades between these teams, with multiple players having played for both sides. Indeed, the presence of a complement mutation may not become significant without the stress of a clinical trigger. An otherwise strong thrombotic thrombocytopenic purpura (TTP) team missed the cut for the NephMadness bracket because of its underwhelming effect on the kidneys.

Figure 1.
When should you suspect TMA?
You are admitting a patient with AKI, proteinuria, and hematuria. You notice thrombocytopenia, anemia, and an elevated reticulocyte count. Could this be microangiopathic hemolytic anemia (MAHA)? You look for elevated lactate dehydrogenase (LDH), low haptoglobin, schistocytes on the peripheral smear, and increased reticulocytes. Check! Direct antiglobulin (or Coombs test) is negative, excluding autoimmune hemolysis. Coagulation parameters are normal, excluding disseminated intravascular coagulation (DIC). Yes, this is the classic presentation of TMA. However, any or all of the MAHA features can be absent, especially in renal-limited disease, and this is when a high index of suspicion is called for. The presence or absence of schistocytes is a particularly debatable disease marker.
Can biopsy help?
Kidney biopsy is not a requisite when distinct hematologic features of MAHA are in plain view, as histopathology is unlikely to supply the exact cause of TMA. Moreover, thrombocytopenia and anemia can sometimes make the biopsy a risky game. In classic cases, successful empiric treatment itself would clinch the diagnosis. On the other hand, it’s been observed that arterial wall changes are more specific for malignant hypertension (HTN) or scleroderma renal crisis, whereas glomerular changes are more specific for CM-TMA. In addition, renal-limited disease can only be diagnosed by pathology. Therefore, a biopsy should be considered when the clinical picture is ambiguous, there is no response to definitive therapy, the degree of reversibility of kidney injury is unclear, or a second pathology is suspected.
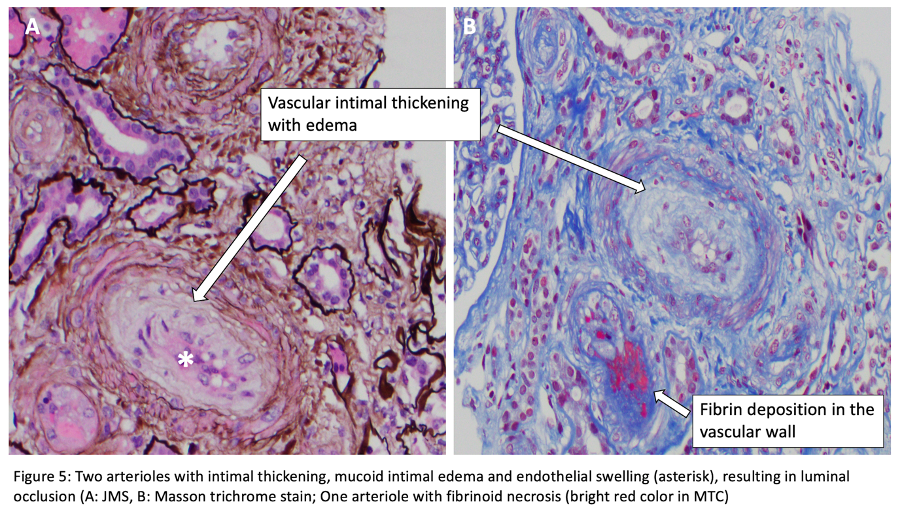
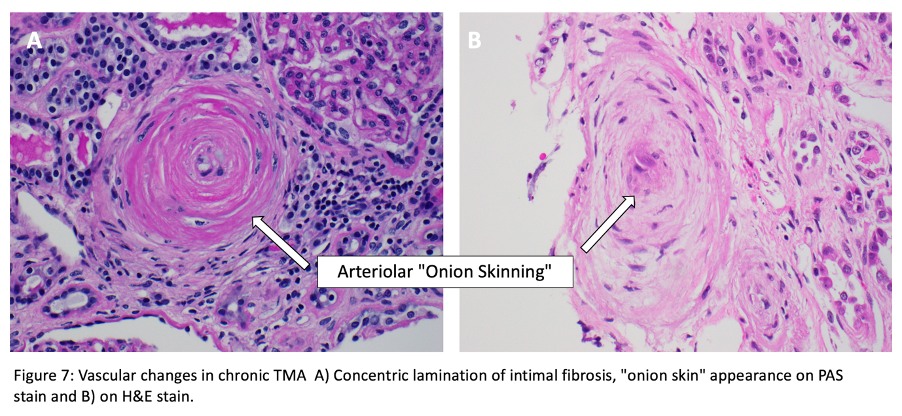
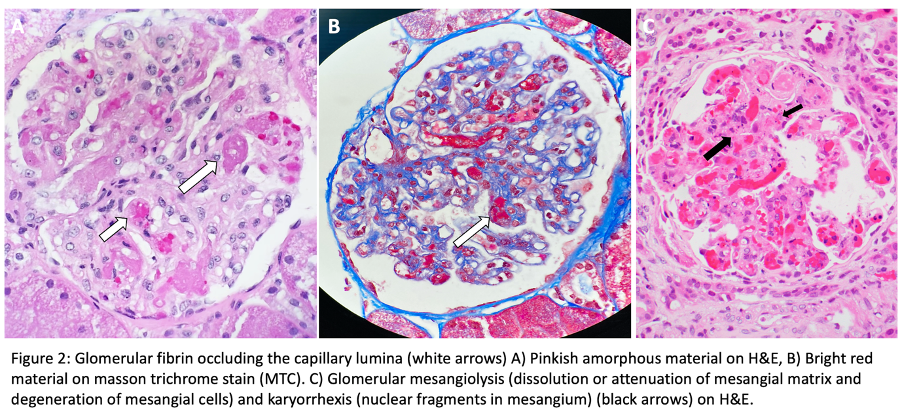
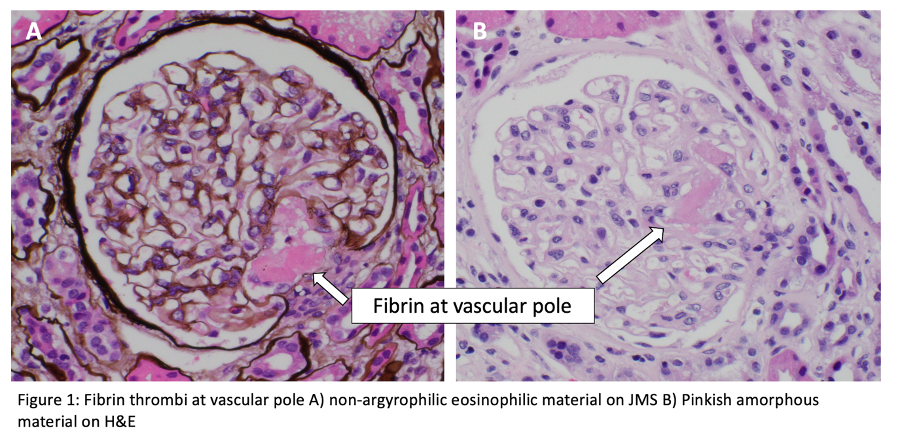
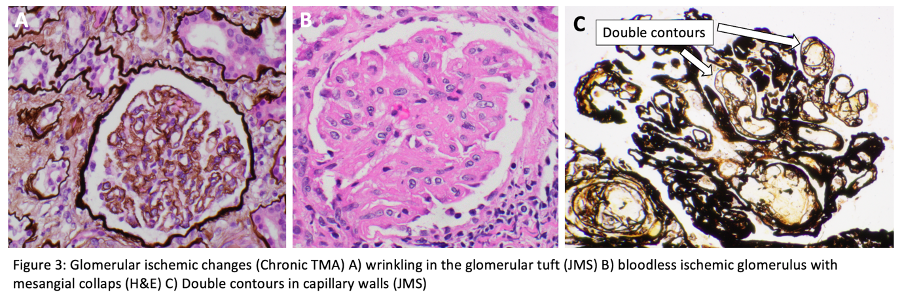
For histopathologic manifestations of renal TMA, we would like to refer fans to this excellent Renal Fellow Network post. Acute vascular lesions associated with such TMAs as malignant HTN or scleroderma showcase mucoid intimal edema and fibrin deposition, while chronic recurrent injury boasts concentric myointimal proliferation (onion-skinning). In the glomeruli, the telltale lesions are microthrombi (fibrin-platelet plugs) in the capillaries or vascular pole. Acute cases might also flaunt endotheliosis (swelling of endothelial cells) and mesangiolysis (loosening of the mesangium), while double GBM contours and segmental glomerulosclerosis are signs of chronicity. As mentioned, histopathologic evidence of TMA can be a surprise finding in a number of glomerular and autoimmune diseases, without the hematologic features.

Copyright: RaiDztor/Shutterstock
Team 1: Primary TMA
“An alternative way
To find a complement
Can be quite quirky!”
Haiku by app/writeme.ai<a id=”Primary”></a>
“…Alice soon came to the conclusion that it was a very difficult game indeed.”
Lewis Carroll, Alice’s Adventures in Wonderland
Alternative Pathway of Complement Activation
Complement-mediated TMA (CM-TMA) results from dysregulation of the complement system, mainly the alternative pathway, which then leads to inappropriate activation of the terminal pathway. In Primary TMA, alternative pathway dysregulation can be genetic (due to mutations in complement proteins), acquired (due to autoantibodies against complement proteins), or idiopathic. The alternative pathway was an MVP in NephMadness 2019 for the Complement Region. To refresh our memory, let’s play a game of “Alternative Basketball” (Figure 2). Initially, the teams are Self vs Pathogens. On the Self team, alternative pathway complement activation is the “offense”, alternative pathway complement regulation is the “defense”, and formation of C3 convertase is the “basket”.
The game starts with a jump ball, C3. The alternative pathway is constitutively active due to spontaneous hydrolysis of small amounts of C3, the so-called C3 tick-over. This auto-activated C3 will deposit on pathogens. To score a point, the “offense” (two proteases, Factors B and D, and a stabilizing protein, Properdin [P]) will engage with C3 basketball to create the powerful alternative pathway C3 convertase. Once C3 convertase is formed, a series of slam dunks via an efficient amplification loop generates large amounts of C3b on the surface of the pathogen. This will opsonize the hapless pathogen and activate the terminal pathway with its lethal weapon, membrane attack complex (MAC) C5b-9.
Following the game so far? The problem is, the auto-activated C3 will also deposit on self-cells. The offense will go rogue and try to score points by threatening cells on its own Self team! Our body must keep a strong defense to avoid damage from our own complement system. This defense consists of plasma proteins (Factors H and I) and membrane proteins (cell defense players CD46/MCP [membrane cofactor protein], CD35/CR1 [complement receptor 1], CD55/DAF [decay accelerating factor] and CD59/MAC-IP [MAC-inhibitory protein]). One example of defensive play is when either Factor H or MCP holds down C3b, allowing serine protease Factor I to inactivate it. A normal complement system, an all-star basketball team, is finely tuned and prepared to deal with any offense, even coming from within. In CM-TMA, weak defense (due to loss of function of regulators) or excessive offense (due to gain of function of complement components) will end up with a series of “self-baskets”. The Pathogen team can sit on the sidelines and cheer as the game turns into Self vs Self!

Figure 2. Alternative Basketball.The complement is on the Self team, and its offense against pathogens consists of factors B, D and P (depicted by player BaD Pee), propelling hydrolyzed C3 (basketball) through the C3 convertase (basket) to produce C3b (to score). Before you know it, the amplification loop repeats this multiple times, producing tons of C3b. However, offense can also go rogue against self cells. To contain its offense, the Self team has a robust complement defense, consisting of factors H and I in plasma and CD 46/MCP, CD55, CD35 and CD 59 on cell surface. This “alternative” version of basketball is the real game played by the complement alternative pathway inside our body every day!
Genetic CM-TMA
The etiology of genetic CM-TMA is typically a heterozygous, loss-of-function mutation in a defense protein of the alternative pathway. Such mutations lead to haplo-insufficiency (the protein is either not made or, if made, does not function properly) and weaken the defense. Less frequently, a gain-of-function mutation in a complement activator is identified. Approximately 60% of patients with CM-TMA carry a rare genetic variant in one of the five complement proteins playing our game (Figure 2):
Loss-of-function mutation of complement Factor H
Loss-of-function mutation of complement Factor I
Loss-of-function mutation of CD46/MCP
Gain-of-function mutation of C3
Gain-of-function mutation of complement Factor B
Of note, approximately 10% of affected patients carry more than one variant. Disease penetrance is approximately 50%, therefore a precipitating event (ie, infection in children, pregnancy in adults) may be necessary to manifest the disease.
If not identified and treated in a timely manner, prognosis is the worst in patients with Factor H mutations, with 60%-70% reaching kidney failure within a year of disease onset. The prognosis in patients with Factor I and C3 mutations is also poor. Patients with CD46 mutations have a better prognosis, with approximately 80% remaining dialysis-independent.
Acquired CM-TMA
Acquired deficiency due to autoantibodies against Factor H is observed in approximately 5%-10% of patients with CM-TMA. A deletion in CFHR1 and CFHR3 (Complement Factor H-related 1 and 3) may indicate a genetic predisposition to the development of Factor H autoantibodies. Autoimmune diseases, usually playing for the Secondary TMA team, can switch sides to cause this entity.
Workup of Primary TMA
How does one join the CM-TMA team? Coaches and scouts (or you, our reader) can use several tests to evaluate the presence of functional complement dysregulation in addition to testing for genetic complement mutations (Figure 3).

Figure 3. From New Insights into TMA in Kidney Transplantation in: Kidney News Volume 14 Issue 8 (2022), by Anuja Java.
The mutations reported as “pathogenic” or “likely pathogenic” are known to cause dysregulation of complement activation. However, less than 50% of the genetically identified variants have a known functional consequence. Computational prediction algorithms are used to predict the potential impact of these “variants of uncertain clinical significance” (VUS) on the mature protein. The presence of such variants is particularly vexing for clinical management. Functional analysis of VUSs can assist in defining the significance of the variant and helping to re-classify them as pathogenic or benign.
Treatment of Primary TMA
Terminal pathway blockade is the best therapy at this time. Two C5 inhibitors, eculizumab and ravulizumab, administered as infusions, have greatly improved disease outcomes with extensive clinical trial- and real-world-evidence of efficacy and safety. Studies are on the way with another C5 blocker, crovalimab, which should simplify therapy with subcutaneous injection. Alternative pathway inhibitors in development (see illustration) are pegcetacoplan (anti-C3), iptacopan (anti-factor B) and danicopan (anti-factor D).
Acquired CM-TMA secondary to Factor H autoantibodies often responds to plasma exchange and immunosuppression.
Liver transplantation can be curative in genetic CM-TMA caused by mutations since a majority of the complement regulatory proteins are of hepatic origin. Therefore, combined liver-kidney transplant may be a feasible option for kidney failure in younger patients, though not commonly used due to the effectiveness of anti-complement therapy. A concern that liver allograft may not initially produce sufficient amounts of the regulatory factors may be circumvented by plasmapheresis in the perioperative time to restore levels of the complement proteins.
COMMENTARY BY JOHN SPERATI:
It’s Always Rosy in the World of Complement
Check out this podcast episode of Freely Filtered featuring Anna Vinnakova and Anuja Java:
#57 NephMadness 2023 TMA

Copyright: Amelia Martin/Shutterstock
Team 2: Secondary TMA
“Of secondary thrombotic
microangiopathy beware.
It’s a tricky affair!
Haiku by app/writeme.ai
Secondary TMA is associated with specific disorders. This roster deploys a diverse set of skills. So what do all these players have in common? The answer is a multi-hit endothelial injury, which renders endothelium vulnerable in the absence of concerted action of plasma and membrane-bound complement regulators. If this team is able to outwork the Primary TMA team (harboring complement mutations) they may be able to exploit certain players (eg, pregnancy, malignant HTN etc.), and generalized complement activation can ensue.
Secondary TMAs are best managed by treating the underlying causes. Or are they?
Evaluation of Secondary TMA

Figure 4. When secondary TMA is suspected, the following tests will help with the differential diagnosis.
Hypertension-associated TMA
Features of TMA can be observed in up to 40% of patients with malignant hypertension, viewed as the proverbial vascular damage from shear stress. Patients with clear precipitants such as cocaine use usually respond to antihypertensives with quick resolution of TMA. However, there is growing evidence that complement activation is the underlying disorder behind the majority of malignant hypertension cases, particularly in young patients. This player might soon be switching teams! Genetic testing is recommended in all patients with recurrent episodes of hypertensive emergency, TMA, and progressive kidney disease. Such patients have high rates of progression to kidney failure and disease recurrence after transplantation. Complement blockers may improve clinical outcomes. In a large study by Cavero et.al (Figure 5), 20% of the patients with malignant hypertension had TMA, and 60% of those were due to aHUS. Over 70% ultimately required dialysis, much worse than those without TMA.

In a small earlier study of 26 patients with hypertensive emergency and TMA on kidney biopsy, 69% had evidence of complement activation, and half had pathogenic variants in complement genes. C5 inhibition successfully restored kidney function in 83% of patients with evidence of complement activation, while HTN control did not make a difference.
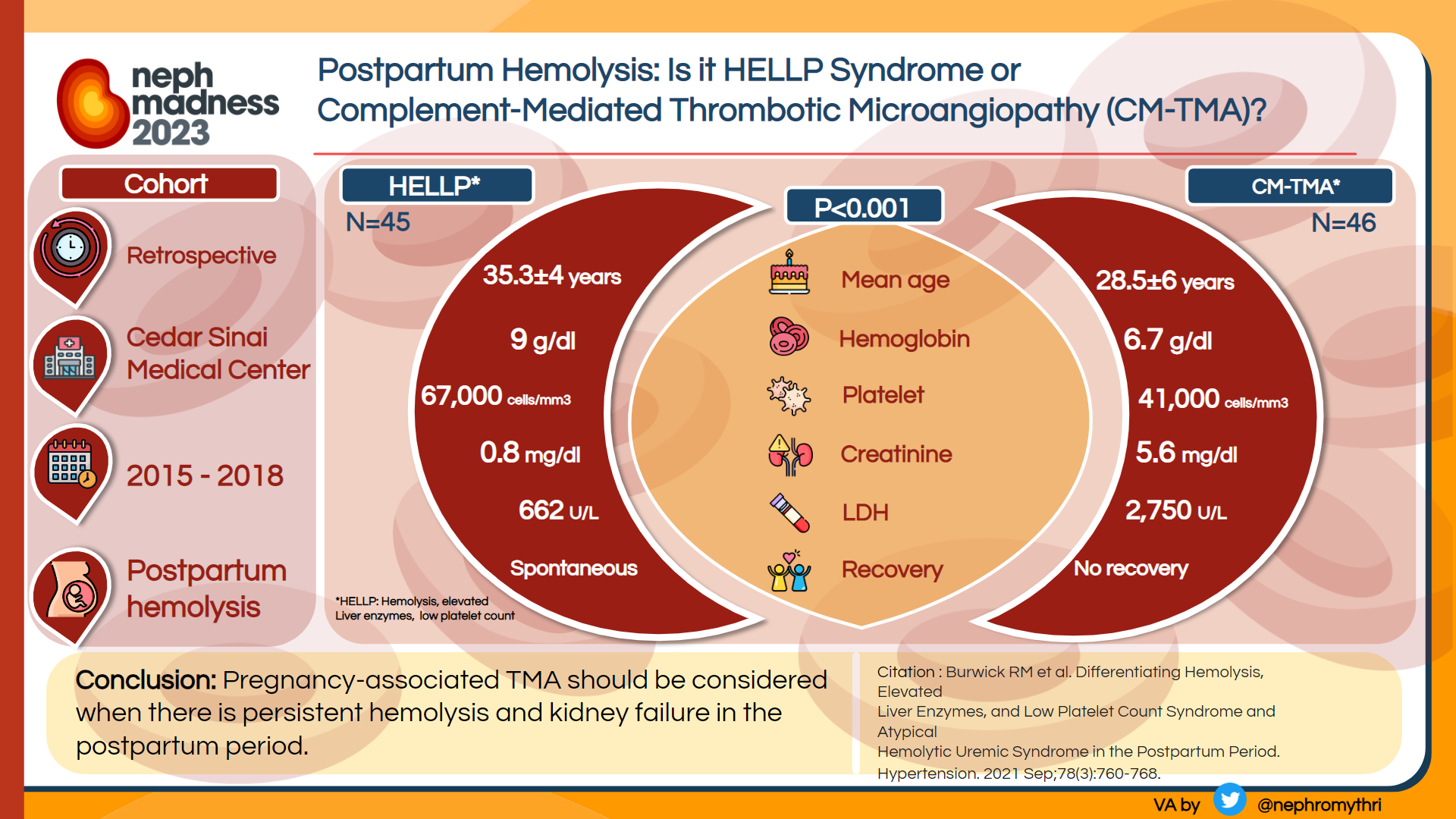
Pregnancy-associated TMA
This team features a “big 3” in stars, Preeclampsia/HELLP (Hemolysis, Elevated Liver enzymes and Low Platelets), TTP, and CM-TMA. Telling these triplets apart is a challenge even without factoring in players like Catastrophic Antiphospholipid Syndrome or Renal Cortical Necrosis with DIC.
Preeclampsia results from abnormal placentation with production of anti-angiogenic (i.e., anti-VEGF) factors triggering complement activation. This typically occurs later in the pregnancy; helpful diagnostic tests are abnormal uterine doppler and elevated sFlt-1/PlGF ratios (the latter are not yet available in the United States). Failure of TMA to resolve upon delivery is suggestive of other etiologies.
TTP is diagnosed by ADAMTS13 deficiency and responds to plasma exchange (congenital TTP) or immunosuppression (immune-mediated TTP). In unusually severe cases of pregnancy-associated TMA, especially those that fail to improve after delivery, plasma exchange should be started empirically until TTP is ruled out.
CM-TMA can be triggered by high risk events late in pregnancy or especially in the postpartum period, and complement gene variants can be found in approximately half of these cases. If untreated, pregnancy outcomes are severe, with two thirds of the patients reaching kidney failure and experiencing a high rate of fetal loss. Recurrence risk in subsequent pregnancies is approximately 25%. Treatment with a C5 inhibitor is associated with improved maternal outcomes without evidence of increased fetal risks. Therefore, patients with undifferentiated pregnancy-associated TMA who fail to respond to plasma exchange and delivery should be started on complement blockade. See Figure 6.
Drug-induced TMA
Drug-induced TMA can be mediated by antibodies or direct toxicity. A comprehensive registry compiled by George et al can help with differential diagnosis.
Immune-mediated drug-induced TMA is due to an antibody (Ab) formation against a drug or its metabolite. This team’s MVP, quinine, an alkaloid derived from the bark of cinchona trees, can stimulate Ab cross-reactive with platelets. Ever since its fall from grace for dabbing in drug induced-TMA, some US nephrologists are nostalgic for the days when this antimalarial agent was used as a remedy for leg cramps. Quinine is responsible for the flavor of gin and tonic and for the fluorescence of glow-in-the dark jello shots. Beware, consumption of these drinks can lead to TMA! Multiple other medications like trimethoprim, ciprofloxacin, metronidazole, and quetiapine can play this game.
Non-immune-mediated drug-induced TMA occurs due to direct drug toxicity to the endothelium by multiple mechanisms. Notorious culprits include calcineurin inhibitors like tacrolimus, chemotherapy drugs like mitomycin C, VEGF inhibitors like bevacizumab, and tyrosine kinase inhibitors like sunitinib. Inappropriate intravenous use of oxymorphone (Opana ER) or oxycodone ER (OxyContin) can lead to drug induced-TMA due to postulated toxicity of polyethylene oxide, a high molecular weight filler intended to make the pills tamper-resistant.
TMA in autoimmune conditions and GNs
Disorders like systemic lupus, antiphospholipid syndrome, systemic sclerosis, C3G, focal segmental glomerulosclerosis, ANCA vasculitis and IgA nephropathy can be associated with TMAs, which portends poor kidney outcome. For example, 80% of patients with systemic lupus erythematosus (SLE) and TMA on kidney biopsy develop kidney failure in 5 years.
Pathogenesis is multifactorial, including fascinating Neutrophil Extracellular Traps, aptly called NETs, akin to basketball nets catching alternative pathway basketballs.
Management is typically geared towards the underlying disorder. Complement blockers may be helpful in select cases, and trials in this disease space are ongoing.
Infection-associated TMA
TMA can be associated with a wide range of viral (HIV, Influenza, COVID19), bacterial, fungal and parasitic infections. The star player, Shiga toxin-producing Escherichia coli-associated hemolytic uremic syndrome (STEC-HUS) was once called the “typical HUS” and wielded more power than the entire “atypical HUS” team. STEC-HUS is most common in children and is associated with Shiga toxin-producing E coli (more severe cases can be caused by Shigella spp). Not to be outdone, Strep pneumo can cause TMA called Strep pneumo-HUS, associated with high morbidity and mortality.
In STEC-HUS, Shiga toxin translocates from the gut, combines with glycolipid Gb3 in the circulation, enters endothelial cells via GB3 receptor, and creates havoc disrupting protein synthesis and egging on tissue factor and von Willebrand factor release. In a fascinating move, Shiga toxin can also bind to Factor H on endothelial surfaces, thus bringing complement into the game. Pathogenesis of Strep pneumo-HUS is attributed to pneumococcal neuraminidase, which cleaves sialic acid residues exposing a cell surface component called T-antigen and initiating complement activation.
Diagnosis is made by stool cultures, PCR testing, and viral serologies. Genetic testing for complement proteins is reserved for severe cases.
Treatment is usually supportive. The role of eculizumab requires further randomized controlled trials.
Malignancy-associated TMA
This form of TMA can be seen with mucin-producing adenocarcinomas as well as disseminated malignancies. What occurs is microvascular obstruction with cancer cells, consumption of platelets in tumor microthrombi, and fragmentation of the passing red blood cells. As we learned in the drug-induced TMA section, chemotherapy agents like mitomycin-C, cisplatin, gemcitabine, or bevacizumab can cause TMA which will typically resolve with the withdrawal of the offending drug.
Eculizumab has been used along with supportive measures in some cases. The mortality rate of cancer-related TMA is high due to its association with disseminated malignancies or withdrawal of chemotherapy.
Transplant-associated TMA
After kidney transplantation, 25%–50% of CM-TMA will recur. In fact, CM-TMA is responsible for most of the recurrent cases and up to a third of the de novo cases. A complement genetic panel is therefore recommended for all patients with transplant-associated TMA. The rest of the cases are broadly caused by the following:
Drugs, such as calcineurin or mTOR inhibitors. The mechanism is direct endothelial injury with release of von Willebrand factor multimers, which overwhelm ADAMTS13 capacity and activate coagulation-complement crosstalk.
Viral infections, such as CMV, Parvovirus B19, SARS-CoV-2. The mechanism is direct endothelial injury by virus with platelet aggregation, generation of thrombin, development of ADAMTS13 inhibitors, and complement activation.
Acute antibody-mediated rejection. This happens via a postulated mechanism wherein a donor-specific antibody (DSA) binds to HLA antigen on endothelial cells and activates complement. Diagnosis of AMR is confirmed by detection of DSA and positive C4d staining on allograft biopsy. AMR-associated TMA harbingers a high risk of graft loss. Transplant glomerulopathy, a chronic smoldering form of TMA, can produce MPGN pattern on biopsy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Non-kidney solid organ transplants can rarely develop TMA.
Ischemia-reperfusion, immunosuppressive drugs, acute interfering disease, and a relative deficiency of the von Willebrand factor cleaving protease (ADAMTS13) appear to play a major role in its pathogenesis.
Hematopoietic stem cell transplantation (HSCT) is no stranger to TMA, with an occurrence rate of 3%-39%. A popular 3-hit hypothesis suggests that hit 1 is an underlying predisposition (eg, genetic variant or prior endothelial injury), hit 2 is new endothelial injury (eg, HSCT conditioning regimen), and hit 3 is complement activating triggers described above.
Management of transplant-associated TMA varies from supportive, like withdrawal of the offending agents or use of antivirals, to complement blockade per genetic or clinical evidence.
Metabolism- and coagulation-mediated TMAs and their mimics
These players do not get a lot of court time, but are a force to be reckoned with, as they can mimic or provoke TMA.
Acquired cobalamin deficiency-induced pseudo-TMA from pernicious anemia or malabsorption is as easily missed as it is treated. It mimics TMA like netball does basketball. The hemolysis is not intravascular but intramedullary: macrocytic nucleated RBCs, like over-inflated basketballs, are fragile and susceptible to shredding, especially in the setting of high homocysteine levels.
Pancytopenia, elevated LDH, and peripheral schistocytes can all be seen. There is an absence of the usual degree of endothelial dysfunction or platelet microthrombi. AKI, if it happens, is due to ATN/pigment injury. The best clue to this pseudo-TMA is reticulocytopenia, while low B12 with high MMA and homocysteine levels are diagnostic. The best therapeutic approach is repletion of Vitamin B12, folate, CoQ-10, and carnitine.
Methylmalonic aciduria and homocystinuria, cobalamin C type (MMACHC), or simply cobalamin C disease, is ultra-rare, but is the most common genetic type of functional B12 deficiency. Genetic testing should be considered in all young patients with evidence of intravascular hemolysis, even in the absence of concomitant renal and neurological impairment. B12 levels are notoriously unreliable unless accompanied by elevated homocysteine and MMA. Treatment with hydroxocobalamin and betaine is effective in preventing progression of kidney disease and recurrent TMA.
Diacylglycerol Kinase ε (DGKE)-TMA manifests in infants with progressive CKD and kidney failure. Loss-of-function DGKE mutation can cause endothelial damage with activation of protein kinase C, upregulation of prothrombotic factors, downregulation of the VEGF receptor, podocyte injury, and TMA. There is insufficient data to determine optimal management.
Variants in Thrombomodulin (THBD), a transmembrane glycoprotein with anticoagulant properties and a negative regulator of the complement system on vascular endothelial cells, may lead to a TMA.
Plasminogen (PLG) is a circulating glycoprotein known to hinder platelet aggregation through its protease form, plasmin. PLG variants can decrease proteolytic activity of plasmin and trigger TMA.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked recessive condition that occurs due to mutations in G6PD, and is another TMA mimic. Patients typically present with hemolysis after exposure to an infection or drug, sometimes accompanied by thrombocytopenia and ATN. Finding G6PD deficiency by measuring G6PD activity in red blood cells once TMA is in remission can spare a patient with TMA the risk and cost of long-term complement blockade.
– Executive Team Members for this region: Anna Vinnikova @KidneyWars and Timothy Yau @Maximal_Change
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC per region through NKF PERC (detailed instructions here). The CME and MOC activity will expire on June 1, 2023.
Submit your picks! | #NephMadness | @NephMadness | #TMAregion
![]()
Leave a Reply