
NephMadness 2016: Transplant Nephrology Science Region

Submit your picks! | For more on NephMadness 2016 | #NephMadness or #TransplantRegion on Twitter
The Transplant Nephrology Science region is full of potential. The region is comprised of exciting teams but unproven on the big stage. Will one of these diaper dandies make the breakthrough in NephMadness ’16 or is this a season too early for our transplant great hopes? The top of the region features a game between improving matching with Team HLA Epitope Matching versus Tregs in transplant. The bottom half of the regions could prove to be a game changer in diagnosing allograft pathology in Team Omic versus the most challenging issue in transplant (what is the immune status of your patient) in Team ELISPOT. All of these matchups could revolutionize kidney transplantation. Do they have what it takes to make a deep run in NephMadenss ’16. We will see.
Selection Committee member for the Transplant Nephrology Region:
Milagros (Millie) Samaniego, MD
Dr. Samaniego is Professor of Medicine in the Division of Nephrology and Medical Director of Kidney Transplantation and Transplant Outpatient Services at the University of Michigan. Dr. Samaniego conducts research in transplantation of highly sensitized kidney transplant recipients and antibody- and complement-induced endothelial cell injury. Dr. Samaniego is an active member of the American Society of Nephrology, the American Society of Transplantation, and the National Kidney Foundation. She is KDIGO co-Chair of Public Policy and editorial member of Advances in Chronic Kidney Disease.

Meet the Competitors for the Transplant Nephrology Region
HLA Epitope Matching
Tregs in Transplant
Omics for Allograft Pathology
ELISPOT for Immune Function
HLA Epitope Matching vs Tregs in Transplant
HLA Epitope Matching
 Matching donor allografts to recipients using HLA profiles proved to be useful mechanism to improve allograft survival in the early years of kidney transplantation. However, in the era of more potent immunosuppression, this beneficial effect has been largely, although not completely, abrogated. Certainly, zero-mismatched allografts (at A, B, DR) do better but even well-matched transplant recipients may form anti-HLA antibodies and develop immunological injury.
Matching donor allografts to recipients using HLA profiles proved to be useful mechanism to improve allograft survival in the early years of kidney transplantation. However, in the era of more potent immunosuppression, this beneficial effect has been largely, although not completely, abrogated. Certainly, zero-mismatched allografts (at A, B, DR) do better but even well-matched transplant recipients may form anti-HLA antibodies and develop immunological injury.
Traditional HLA matching includes only HLA-A, B, and DR antigens and also ignores the immunogenicity of the antigen mismatch (the ability to induce an antibody response). The immunogenicity of a particular HLA antigen mismatch depends on the amino acid structure of both the recipient and donor antigen and specifically the level of dissimilarity between the two. HLA antigens contain many strings of polymorphic sites (epitopes) that serve as targets for antibody binding. Rather than binding to the whole antigens, antibody binds to epitopes, specifically epitopes not present on self-HLA antigens.
Each structural epitope contains a functional epitope of 2–5 residues, some of which are non-self, that determine the strength and specificity of antibody binding. The term “eplet” is used to describe patches of these polymorphic residues in close proximity to each other. Defining HLA at the eplet level allows for a more detailed assessment of HLA compatibility.
It increasingly becoming apparent that late kidney loss is largely an immunologically mediated event and that anti-HLA antibodies (particularly donor-specific antibodies [DSA]) are usually the culprit. Therefore, our current crude assessment of “matching” — which has not changed for decades — is due for an overhaul. This is where Team HLA Epitope Matching comes into the fray and helps explain why some mismatches are permissive (no DSA formation despite antigen “mismatch”) and other are not.
Computer programs are available which can define the number of epitope mismatches for particular HLA antigens (e.g., HLA Matchmaker). The theory is that the higher degree of shared epitopes between donor and recipients, the lower the odds of developing DSA, and vice versa. So does this hold true? Wiebe et al employed HLA Matchmaker to characterize Class II HLA epitope mismatches in 286 consecutive donor/recipient pairs. They defined a threshold for epitope mismatches (10 for HLA-DR, 17 for HLA-DQ) that was associated with development of Class II de novo DSA. Moreover, a study published this month describes the influence of physicochemical properties of the amino acid combinations (determined by the polar charges of amino acids) in HLA alloantigen immunogenicity. This is in addition to the actual amino acid or eplet mismatch described above. HLA matching is getting a lot more complicated.
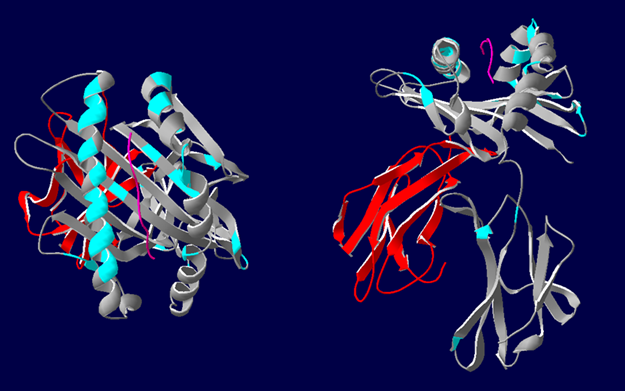
3D representations of HLA-A*02 showing the polymorphic regions accessible for antibody binding. β2-microglobulin (red), HLA class I α-chain (grey) with epitopes highlighted in blue. Most polymorphic regions are focused around the peptide (pink). Image courtesy of Richard Battle, Histocompatability & Immunogenetics Laboratory, Royal Infirmary of Edinburgh. Created using HLA Matchmaker.
Other ways in which functional epitope (eplet) mismatching may be useful:
- To help us understand otherwise unexplained sensitization patterns induced by given mismatch (shared epitopes across different antigens). For example, sensitization to certain HLA-C mismatches may produce antibodies cross-reacting with epitopes shared with HLA-B antigens. This phenomenon has also been reported for Class II antigens, specifically between certain HLA-DR subtypes.
- Acceptable mismatch programs. Eurotransplant has incorporated epitope analysis in their program to improve definition of matching donor and highly sensitized recipients.
- To determine immunogenicity of a particular mismatch in a particular recipient via epitope load. Certain antigen mismatches may be tolerated structurally as there are few epitope mismatches where in another recipient it may be associated with a high burden of epitope mismatches.
- To improve HLA-DP matching in re-transplants. It has been noted that exposure to donor-specific DP antigens can cause generation of anti-DP antibodies which are both donor and non-donor specific. These antibodies however, recognize shared epitopes across the unrelated HLA-DP molecules. This concept is also demonstrated in this report of shared epitopes across different DP antigens in a first and third transplant causing ABMR in the third transplant due to pathogenic DSA.
The burning question here is will this approach improve outcome? The data are conflicting and need to be updated for the current era. Analysis of UNOS and Eurotransplant kidneys transplanted from 1987 to 1999 demonstrated equivalent graft survival between HLA-A and HLA-B mismatched kidneys with low epitope loads and zero HLA-A and HLA-B antigen mismatched kidneys. However, analysis of HLA-A and HLA-B mismatched kidneys from the Collaborative Transplant Study between 1991 and 2001 showed no benefit in graft survival by examining epitope matching. Another Collaborative Transplant Study analysis, this time based on HLA-DPB epitope matching, reported certain epitopes responsible for HLA-DPB immunogenicity, with those harboring <2 HLA-DPB epitope mismatches having a significantly better graft outcome.
So the jury may be out for now on HLA matching at an epitope/eplet level. It’s an exciting young prospect with a body of support behind it. However, it requires clarification of benefit in modern studies for hard outcomes like graft survival before the community at large gets behind it.
Tregs in Transplant
 Determining if an allograft is accepted or rejected depends on the balance between suppression of effector T cells and the promotion of regulatory T cells (Tregs). T cells have fluidity in their life cycle and in certain circumstances can alternate between pro- and anti-inflammatory phenotypes depending on their environment. Most research in this area concerns CD4+ T cells which express the self-regulatory transcription factor Foxp3. In basal conditions, these Tregs account for 5-10% of total CD4+ T cell numbers. They function in immune recognition and can suppress pathogenic effector T cells. They are important in antitumor and anti-infection activity and autoimmunity, with loss of function mutations in Foxp3 causing a devastating autoimmune syndrome. They are also crucial for self-recognition and therefore of paramount importance in achieving the holy grail of transplantation, allograft tolerance.
Determining if an allograft is accepted or rejected depends on the balance between suppression of effector T cells and the promotion of regulatory T cells (Tregs). T cells have fluidity in their life cycle and in certain circumstances can alternate between pro- and anti-inflammatory phenotypes depending on their environment. Most research in this area concerns CD4+ T cells which express the self-regulatory transcription factor Foxp3. In basal conditions, these Tregs account for 5-10% of total CD4+ T cell numbers. They function in immune recognition and can suppress pathogenic effector T cells. They are important in antitumor and anti-infection activity and autoimmunity, with loss of function mutations in Foxp3 causing a devastating autoimmune syndrome. They are also crucial for self-recognition and therefore of paramount importance in achieving the holy grail of transplantation, allograft tolerance.
Adding to the complexity of the Treg system is the fact that many subtypes of Tregs exist, with nTregs (natural; developed in thymus) and iTregs (induced; converted in the periphery from effector T cells) being 2 broad categories. These cell types have different roles such as antigen specificity and also differ in the conditions which will induce or suppress them. Put simply, nTregs may have more of an auto-reactivity role whereas iTregs may be more involved in reactivity to foreign antigens. A nicely pitched review for the general nephrologist is available on Medscape.
So how may Tregs be of clinical benefit in transplantation?
- Improved diagnostics? Foxp3 expression on allograft biopsies has been investigated as marker of immune activity but its relationship with transplant prognosis has demonstrated conflicting results at times. This is possibly due to the fact that Foxp3 can be expressed by both recently activated effector T cells and as well as Tregs. Therefore, more work is needed to clarify its role in graft histology, if any. The possibility of using urine Foxp3 expression should also be explored (higher expression found in operationally tolerant patients) including the methylation status of Foxp3 — i.e., is the gene silenced or not? (see NephMadness 2015-epigenetics)
- Treatment of rejection? Could administration of donor specific Tregs be used therapeutically in acute rejection? This remains purely speculative at this point but is an intriguing prospect. There is real-world evidence that following ATG induction, there is an expansion of Tregs and that following basiliximab induction, a sustained high Treg/effector T cell ratio is associated with the absence of acute rejection. However, data for therapeutic administration of Tregs during a rejection episode are lacking. A pilot study is planned (to recruit 3 patients!) with inflammation on a 6 month surveillance biopsy and administer Tregs with a view to assessing the safety profile and ultimately to preserve allograft function. Another ongoing trial in the UK plans to administer nTregs to reduce acute rejection risk in living donors.
- Induction of tolerance? The hope for tolerizing protocols in transplantation is to induce prominent iTregs that confer donor specific tolerance. In a mouse model of bone marrow and subsequent skin and cardiac allografts, Tregs stimulated for both directly and indirectly presented alloantigens were administered (see image). They prevented both acute and chronic rejection, with Tregs stimulated by directly presented donor antigens only preventing acute rejection. This was, however, in the context of significant pre-transplant irradiation.

Credit: Figure 2 from Hricik AJKD; reproduced with permission of NKF.
There is an ongoing human study involving infusion of Tregs post-transplant in live donor kidney transplants. The Tregs will be harvested from a mixture of recipient peripheral blood mononuclear cells (PBMC) stimulated with donor PBMC in the presence of the costimulatory blocker belatacept. Belatacept is an interesting choice in this study because indirect antigen presentation, as occurs with host antigen presenting cells, may be the key process of allopeptide-induced Treg or effector T cell expansion.
The alternate approach would be to expand Tregs in vivo. Potential mechanisms may include manipulation of Foxp3+ Tregs via cytokines or other immunosuppressive agents and epigenetic modifications (methylation/demethylation are off/on switches for the Foxp3 locus). Data with immunosuppressive therapy on Treg profiles are interesting and divergent. mTOR inhibitors have consistently demonstrated a more favorable Foxp3+ Treg profile to calcineurin inhibitors (CNI). Despite this, clinical studies have consistently demonstrated their inferiority to CNI, with the ELITE-Symphony study being the most high-profile example of this. Much remains to be learned in the complex relationship between drug therapy and T cell function.
Induction of tolerance using iTregs has been hampered by the complexity of the system, including the competition between nTregs and iTregs in vivo and the phenomenon of fluidity mentioned above. This results in Tregs losing Foxp3 expression, with a resultant loss of immunosuppressive function and potentially a gain of harmful effector T cell function, depending on the inflammatory milieu they find themselves in.
Various protocols for inducing tolerance exist, mostly in HLA-identical transplants, with Stanford, MGH, and Northwestern being leading centers currently conducting trials. Pre-conditioning usually involves anti-lymphocyte preparations and lymphoid irradiation. Withdrawal of immunosuppression is conducted over time, based partly on the presence of durable donor chimerism. Potential adverse effects include “engraftment syndrome”, characterized by severe but temporary renal dysfunction and graft versus host disease. Tregs are induced either by concomitant same-donor bone marrow transplant or administering an engineered mix of tolerogenic “facilitating cells” (mixture of precursor dendritic cells and CD8+ T cells not expressing T cell receptors). When chimerism is successful, recipient T cells have show donor unresponsiveness, characterized by abundant Treg signatures, and high levels of intragraft Foxp3 expression. Results have been mixed, with some patients remaining free from immunosuppression and appearing tolerant. Others have lost chimerism, with some remaining hyporesponsive to the allograft and others developing immunological injury.
While we have focused on T cells here, where the wealth of data lies, B cells are also crucial for achieving tolerance. They are obviously potent antigen presenters for alloreactive effector T cells and their signals may facilitate Treg generation. B cell depletion with subsequent manipulation of the re-emergent cell line to negatively select alloreactive clones is being attempted using anti-BLyS (B cell survival factor) therapy.
Attaining allograft tolerance by either direct administration of Tregs or inducing Tregs is a tantalizing goal, based on solid theory but proving very difficult to achieve with realistic protocols. Inducing tolerance in HLA-identical living donor transplants, a group with already excellent allograft survival, is perhaps not all that interesting and the real test will be to make tolerance achievable for all transplants, including for HLA-incompatible pairs. This is a team with big name players with a formidable reputation. It is time for it to deliver in the big dance.
Omics for Allograft Pathology vs ELISPOT for Immune Function
Omics for Allograft Pathology
 We rely on blunt instruments to diagnose allograft pathology — serum creatinine, proteinuria, and visual assessment of graft histology. Even newer tool such as the presence of DSA and C4d staining of biopsies are inconclusive as standalone tests, with an appreciable false-positive and false-negative rates. Employing “omics” technology to describe what is actually happening in the kidney tissue at a molecular level holds great promise to achieve precise diagnosis in renal transplantation. This work has been pioneered by Phil Halloran’s laboratory in Alberta, Canada. These studies have involved microarrays and other gene expression assays to define molecular signatures associated with classic histological signs. Work was initially performed using animal models and subsequently in human real-world indication biopsies, including multiple validation cohorts. These “pathogenesis-based transcript sets” (PBTs) appear to segregate based on the rejection phenotype:
We rely on blunt instruments to diagnose allograft pathology — serum creatinine, proteinuria, and visual assessment of graft histology. Even newer tool such as the presence of DSA and C4d staining of biopsies are inconclusive as standalone tests, with an appreciable false-positive and false-negative rates. Employing “omics” technology to describe what is actually happening in the kidney tissue at a molecular level holds great promise to achieve precise diagnosis in renal transplantation. This work has been pioneered by Phil Halloran’s laboratory in Alberta, Canada. These studies have involved microarrays and other gene expression assays to define molecular signatures associated with classic histological signs. Work was initially performed using animal models and subsequently in human real-world indication biopsies, including multiple validation cohorts. These “pathogenesis-based transcript sets” (PBTs) appear to segregate based on the rejection phenotype:
- T Cell–Mediated Rejection (TCMR). Primed effector T cells cross capillary membranes, interacting with antigen-presenting cells (APCs) carrying donor antigen, and leading to characteristic inflammation. Top TCMR transcripts reflect activated effector T cells, APCs, and CTLA4 co-stimulation. Using these approaches it appears that TCMR is uncommon after 5 years post-transplant and exceedingly rare after 10 years.
- Antibody-Mediated Rejection (ABMR) phenotypes:
- Type 1: early onset in sensitized patient; predictable.
- Type 2: later onset frequently in non-sensitized non-adherent patients; TCMR frequently co-exists; Class II HLA antigens commonly predominate.
- Transplant glomerulopathy is often a late stage of the ABMR process, but some cases are DSA negative and have no indication of antibody-induced injury. Histology is reflected by basement membrane double contours and a high Banff chronic glomerulopathy (cg) score.
The top transcripts for ABMR-related phenotype reflect angiogenesis and endothelial injury in the microcirculation including NK cells transcripts. Some crossover exists between TCMR and ABMR transcripts. The INTERCOM study validated the above classifiers prospectively in a distinct multinational transplant cohort undergoing indication biopsies.
The ABMR score may be particularly useful in cases where uncertainty exists (for example C4d-negative biopsies) and to distinguish chronic ABMR from non-rejection “atrophy-fibrosis” in transplant glomerulopathy patterns of injury. A recent study from the Halloran group further characterizes the molecular sub-phenotypes of an ABMR cohort into pgABMR, pgcgABMR, and cgABMR lesions. This was based on the presence or absence of peritubular capillaritis or glomerulitis scores (p+g) and cg scores. Higher cg score cases were more likely to occur later, to occur without DSA and with non-adherence underlying. They propose replacing the term “transplant glomerulopathy” with “histologic cgABMR” or “histologic pgcgABMR“.
What are classifiers?
In an effort to remove subjectivity from the diagnosis of allograft pathology, classifiers were developed based on the PBTs. These are machine-learning algorithms that predict the molecular phenotype. The classifiers are trained using the following method:

Adapted from Reeve et al American Journal of Transplantation 2013.
Classifier scores have been shown to correlate well with traditional histological diagnosis (agreement 90% for TCMR; 83% for ABMR) but also re-classify a significant proportion of cases and distinguish TCMR from ABMR (including Type 1 ABMR) from no-rejection.
What about borderline acute rejection?
Inflamed biopsies with <25% of the parenchyma demonstrating lymphocytic infiltration, or low-level tubulitis not meeting criteria for TCMR, may be labelled as “borderline” by the Banff system. This is obviously unsatisfactory for the clinician. It either is or isn’t a rejection but our current tools for diagnosis cannot answer this fundamental question for us. The Halloran group studied 40 borderline, 35 TCMR, and 116 non-rejection biopsies. Their TCMR molecular classifier reassigned the 40 borderline biopsies as TCMR-like in 13 cases (33%) and non-rejection-like in 27 cases (67%). It is notable that the classifier also re-designated 9/35 histological TCMR as molecular non-rejectors. A major cause of discrepancy was the degree of interstitial fibrosis/tubular atrophy (IFTA) which interfered with the assessment of tubulitis and inflammation (as per Banff criteria, the assessment of interstitial inflammation [i-score] must be in non-scarred tissue). The study demonstrates the potential to abandon the ambiguous diagnosis of “borderline TCMR” using molecular techniques.
Some other interesting findings across different studies using a “molecular microscope” include:
- Isolated v lesions: TCMR diagnosed exclusively by intimal arteritis without significant tubulitis or inflammation is a controversial entity. v lesions may occur with ABMR and non-rejection acute kidney injury. Using the TCMR classifier, most diagnoses displayed low TCMR classifier scores.
- Defining ABMR more clearly: The ABMR score could judge probability of ABMR without prior knowledge of histology, C4d, or DSA and was strongly predictive of allograft failure (appeared even more so than conventional assessment). This was also demonstrated in a French cohort using similar ABMR scores.
- Concerns regarding C4d staining: Most late ABMR-score-positive cases were in fact C4d negative and there was no difference in score between C4d-positive & negative cases. Notably, in the prospective INTERCOM cohort, C4d positivity had no prognostic significance over and above the presence of ABMR.
- Fibrosis & Atrophy: A recent JCI paper examined allograft fibrosis (& atrophy) at varying time points post-transplant and showed no fibrosis very early after transplant with a linearly increasing prevalence with time. Early transcripts were in AKI pathways, related to donor-implantation injury and were generally self limiting. Late biopsies expressed ABMR and glomerulonephritis transcripts and also tended to have progressive allograft injury and subsequent failure. The study supports a nephron-centric model of fibrosis caused by continuing injury, rather than autonomous committed fibrogenesis. This suggests that when injury is shut off early, good function may be restored, and vice versa.
A significant benefit that molecular diagnostics bring is to refine our traditional histological diagnosis by using data gathered from molecular signatures. This is important as currently, the “molecular microscope” is a research tool and not available for clinical use. For example, retrospectively, it seems that total i-scores (scarred plus unscarred areas) and an “extent of tubulitis score” (percentage of cortical tubules with tubulitis of any degree) may be more accurate for a diagnosis of TCMR. The real value will likely be in combining molecular and conventional assessment to give improved accuracy, as suggested in the INTERCOM study. As the molecular scores use arbitrary cut-offs (which may be refined as data accrues), some false negatives, and perhaps false positives, will undoubtedly occur. Combined assessment seems to lead to the greatest accuracy in diagnosis.
These studies demonstrate the huge potential for precision diagnostics that molecular assessment can bring to allograft pathology. Despite apparently robust statistical methods, because these tools are based on reference to a flawed gold standard, repeated validation in larger cohorts will be required before they are adopted clinically. The more patients that are included in these studies to refine the classifiers, the more accurate these techniques will hopefully become. Ultimately, these methods should be seen as complementary to traditional histology, which we all admit is imperfect, rather than replacing it.
The molecular microscope has emerged as a real threat to its conference rivals who play a more conventional game. This team has its doubters but with accruing results that are continually positive, this is a team that the Nephrology community are likely to be hearing about for years to come in the tourney.
ELISPOT for Immune Function
 Transplantation medicine is at its core a delicate balancing act between allograft immunological injury and complications of immunosuppression. We over-immunosuppress many, as evidenced by high rates of opportunistic infection and malignancy. We under-immunosuppress many more, demonstrated by rejection phenotypes underlying most early and late transplant failures. We use crude tools to assess immunosuppression burden and immunological risk including leucopenia, drug levels, degree of HLA mismatch, DSA, and allograft histology, an invasive and still subjective test. Therefore, a method of precisely assessing immunological risk would allow for the development of personalized approaches to transplant immunosuppression. Several immune-monitoring assays have been used experimentally to reflect recipient T cell activity.
Transplantation medicine is at its core a delicate balancing act between allograft immunological injury and complications of immunosuppression. We over-immunosuppress many, as evidenced by high rates of opportunistic infection and malignancy. We under-immunosuppress many more, demonstrated by rejection phenotypes underlying most early and late transplant failures. We use crude tools to assess immunosuppression burden and immunological risk including leucopenia, drug levels, degree of HLA mismatch, DSA, and allograft histology, an invasive and still subjective test. Therefore, a method of precisely assessing immunological risk would allow for the development of personalized approaches to transplant immunosuppression. Several immune-monitoring assays have been used experimentally to reflect recipient T cell activity.
Enzyme -linked Immunospot Assay (ELISPOT)
The ELISPOT method had been around for over 3 decades and has been modified for the identification of specific cytokine-producing cells at the single-cell level. Recipient T cells are cultured with inactivated donor or third-party cells in wells coated with a capture antibody specific for the cytokine of interest, frequently interferon
γ.
After culture, the cells are washed away and the bound cytokine is detected (as a “spot” representing a single reactive cell) using a secondary labelled antibody (see image). Digital cameras count up the spots, thereby providing quantitative data. In this way, the ELISPOT assay provides quantification of previous effector or memory T cells that produce interferon γ in response to donor antigens, i.e., a reflection of anti-donor T cell sensitization. The test takes 24 hours (primarily due to the cell culture) meaning it cannot be used for clinical decision making on the night of deceased donor transplant for specific donors. However, using the ELISPOT both before after transplant holds promise to understand the risk of immunological injury.

What about pre-transplant risk assessment?
The ELISPOT test can be performed against a panel of allogeneic stimulator cells expressing multiple HLA molecules, from a representative donor population, to generate a T cell reactivity index. This is analogous to panel reactive antibody (PRA) assays that assess humoral reactivity. There are data suggesting that such a T cell assay may identify patients at risk for TCMR despite the absence of traditional humoral sensitization.
Test of over-immunosuppression:
More recently, T cell reactivity using ELISPOT, in combination with an assay of NK cell function, was studied regarding the development of cancer post-transplant. T cell alloresponse (and NK cell function) was significantly impaired in transplant recipients who had a combined end point of metastatic cancer, cancer-related death, or septic death.
Another potential use is a modified assay to test for infection-specific reactive lymphocytes. Studies are ongoing to determine if this technique is helpful (over and above traditional serological risk stratification) in predicting CMV and VZV infection in a bone marrow transplant population.
Stratification of patients for immunosuppression reduction:
ELISPOT has been studied in recipients undergoing tacrolimus withdrawal as a discriminator between poor and favorable outcomes. The presence of donor-reactive T cell was predictive of de- novo DSA and acute rejection, in combination with the urine chemokine CXCL9 and a high degree of HLA epitope mismatches; urinary CXCL9 has previously been identified as a biomarker of future immune injury in renal transplantation. Overall, however, the outcome with tacrolimus withdrawal was poor so this approach could not be recommended, even using these novel approaches to stratify patients.
What about recipients a long time out from transplant?
Circulating donor-specific alloreactive T cells may be detected in stable long-term renal transplant patients and appears to correlate with allograft injury. This may have implications if immunosuppression minimization is being considered.
Not all the data is positive and in a recent study ELISPOT failed to predict either acute rejection or eGFR post-transplant in a cohort of renal transplant recipients. Subgroup analysis did however show a lower eGFR at 6 & 12 months when donor-reactive T cells were present, but only in those who did not receive ATG induction. This was independent of demographic factors, previous rejection, PRA and conventional HLA mismatch.
Can we use ELISPOT for assessment of humoral immunity?
An ELISPOT assay has been modified to assess B cell response in a renal transplant cohort without detectable DSA using single-bead techniques (n=9). The assay used donor fibroblasts (therefore expressing HLA Class I antigens — remember, these are present on all nucleated cells). It revealed donor reactivity via the increased frequency of donor-specific antibody-secreting cells, indicating that B cells respond to the donor despite DSA not being detectable. This work has helped us understand B cell responses but throws up many questions:
- What are the antibodies directed against? Perhaps HLA not represented by the single antigen beads.
- Why is there no detectable DSA? It could be due to operational tolerance, effects of immunosuppression, or antibody binding to the allograft.
The immune system is a complex beast and no single assay will ever be able to provide a complete picture of the likely reactivity of a patient to a foreign allograft. Could a combination of DSA, ELISPOT, NK cell activity, and perhaps other assays help us more precisely determine immunological risk and tailor immunosuppressive treatment more accurately? Perhaps, but ELISPOT has been around the tournament for many years now and has not yet found its way into the senior divisions. We will have to wait and see if 2016 will be its year.
– Post written and edited by Drs. Paul Phelan and Milagros Samaniego-Picota.
![]()
Leave a Reply