
C4 Nephritic Factor? I Still Don’t Understand C3 Glomerulopathy!
The complement system lies at the interface of innate and adaptive immunity. Perhaps its most remarkable feature is that it reacts in seconds to opsonize and/or lyse a bacterium while simultaneously releasing proinflammatory and chemotactic peptides. The complement system consists of three major activation cascades: the classical, lectin, and alternative pathways.
The classical pathway (CP) is primarily triggered by antibody binding to antigen. In an analogous process, the lectin pathway (LP) is activated by lectin (equivalent to an antibody) binding to a sugar (equivalent to an antigen) on a microbe. After the initial recognition event, the CP and LP feature multiple proteolytic cleavage steps leading to subsequent activation of the components C2 and C4 and the formation of the CP C3 convertase, C4b2ba. This bimolecular enzymatic complex converts C3 into C3a and C3b.
The alternative pathway (AP), however, is continuously activated via the spontaneous hydrolysis of C3 without need for prior contact/exposure. If this auto-activated C3 deposits on healthy self-tissue, host regulatory proteins immediately inactivate it. However, if it deposits on a microbe, activated C3 can be quickly amplified by engaging two proteases, factor B (FB) and factor D, to create the AP C3 convertase, C3bBb. Similar to the CP C3 convertase, this enzyme also converts C3 into C3a and C3b.
A critical feature of the AP is its amplification loop. C3b, no matter how it is generated, serves as a nidus for this feedback loop for the generation of large amounts of C3b to opsonize a pathogen. Addition of C3b to a C3 convertase generates a C5 convertase that cleaves the C5 protein into C5a (a potent anaphylatoxin) and C5b [engages the membrane attack complex (MAC)].
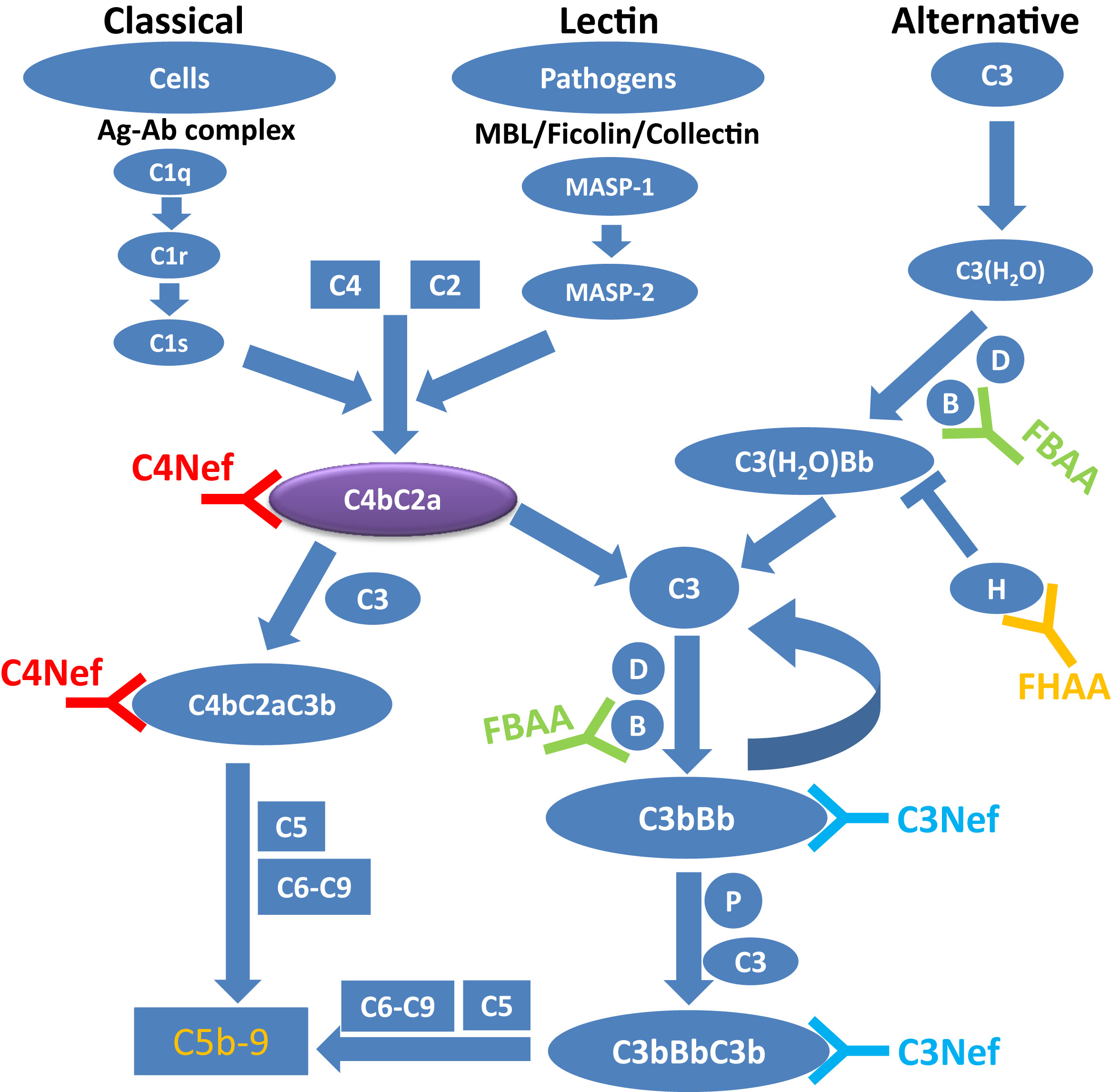
The complement cascade. A simplified schematic of the alternative, lectin, and classical pathways of complement. Each of these initiating pathways leads to the generation of C3 convertases (C3bBb or C4b2a) and C5 convertases (C3bBbC3b or C4b2aC3b). C5 convertases cleave C5 and trigger the generation of membrane attack complex (MAC) or C5b-9. The presence of C4 nephritic factor (C4Nef) and/or C3Nef stabilizes C3 convertases, resulting in hyperactivity of the alternative and terminal pathways. Abbreviations: FBAA, factor B autoantibody; FHAA, factor H autoantibody; MASP, mannose-associated serine protease; P, properdin. Figure 1 from Zhang et al, AJKD © National Kidney Foundation.
Complement activation is designed to eliminate the pathogen. However, if misdirected or dysregulated, it can be similarly damaging to self. Therefore, strict regulation of the complement system is required to prevent collateral damage to host cells and tissues. The regulatory proteins that protect against undesirable AP activation include two in plasma, factor H (FH) and factor I (FI), and three membrane proteins, namely, membrane cofactor protein (MCP), decay-accelerating factor (DAF), and complement receptor 1 (CR1). Inherited defects in complement genes and acquired auto-antibodies against a regulatory protein predispose to several diseases because of inadequate control of complement activation.
C3 glomerulopathy (C3G) is a recently defined entity that encompasses a group of kidney diseases driven by uncontrolled activation of the alternative complement pathway that leads to C3 deposition within the glomerulus. Based on electron microscopy, C3G may be subclassified as dense deposit disease (DDD, sausage-shaped deposits in the GBM) and C3 glomerulonephritis (C3GN, deposits that are discontinuous and less dense). The genetic drivers of disease include rare or novel variants in complement genes, identified in ~ 30-40% of C3G patients, while the acquired drivers include C3 nephritic factors (C3Nefs), FH autoantibodies, and presence of monoclonal gammopathy. Of the acquired factors, the most common and the best studied are C3Nefs reported in up to 80% of patients with DDD and 50% of patients with C3GN. C3Nefs are autoantibodies that bind to and stabilize the AP C3 convertase (C3bBb) by preventing its inactivation by FH. This leads to a prolonged C3 convertase activity resulting in continued conversion of C3 to C3b, which in turn causes consumption of C3.
In a recent AJKD article, Zhang et al (Open Access) sought other potential autoantibodies in patients with C3G who were negative for C3Nef, with a specific focus on C4 nephritic factor (C4Nef). Similar to the C3Nef, C4Nef is a stabilizing antibody to the C3 convertase (C4b2a) of the classical and lectin pathways and prolongs the half-life of the convertase. Very little is known in the literature about C4Nefs, with only five reports in the past nearly 25 years. In these reports, this autoantibody was shown to be associated with postinfectious glomerulonephritis, systemic lupus erythematosus, and meningococcal disease. The last example is particularly interesting because despite an undetectable serum C3 over a 2-year follow-up period, there was no evidence of renal involvement.

C4 nephritic factor (C4NeF) assay. Antibody (Ab)-sensitized sheep erythrocytes (EA) are used as index cells to generate EAC1C4b by incubating pooled normal human serum in the presence of TTHA (a strong magnesium chelator) and calcium. After washes, EAC1C4b2a (C3 convertase of the classical pathway) is made by adding purified C2. To test for the presence of C4b2a autoantibodies (C4Nefs), patient-purified immunoglobulin G (IgG) is added and cells are allowed to decay for different periods (7.5, 15, 22.5, and 30 minutes). After each time point, 50 μL of the cell mixture is removed. An equal volume of rat-EDTA serum (1:19 dilution) is added as a source of complement proteins C3 through C9. To stop the reaction after 1 hour, 150 μL of cooled EDTA-GVB buffer is added. Nonlysed cells are removed by centrifugation and hemoglobin in supernatant (200 μL) is measured at optical density at 415 nm (OD415). Figure 4 from Zhang et al, AJKD © National Kidney Foundation.
Of the 168 biopsy-confirmed C3G patients described in this case series, five (~3%) were found to have C4Nefs. These autoantibodies were absent in 300 normal healthy controls, and not detected in atypical hemolytic uremic syndrome (aHUS), another prototypic complement-mediated renal disease. One of the five patients was also positive for FH autoantibody. No rare or novel genetic variants in complement genes were identified in the C4Nef-positive patients. There was evidence of ongoing complement activation in all patients as indicated by low C3, low C5, and elevated C5b-9. The clinical course was variable with two patients developing chronic kidney disease.
Although it is not entirely clear why particular patients develop C4Nefs, this paper serves as a reminder of the complexity of the autoimmune phenomenon associated with C3G and further underscores the importance of comprehensive complement evaluation in all patients with this disease.
– Post prepared by Anuja Java, AJKDBlog Guest Contributor
To view Zhang et al (Open Access), please visit AJKD.org.
Title: C4 Nephritic Factors in C3 Glomerulopathy: A Case Series
Authors: Y. Zhang, N.C. Meyer, F.C. Fervenza, W. Lau, A. Keenan, G.Cara-Fuentes, D. Shao, A. Akber, V. Fremeaux-Bacchi, S. Sethi, C.M. Nester, and R.J.H. Smit
DOI: 10.1053/j.ajkd.2017.07.004
Leave a Reply