
#NephMadness 2019: Hepatorenal Region
Submit your picks! | NephMadness 2019 | #NephMadness | #HRSRegion
Selection Committee Member: Juan Carlos Velez
Juan Carlos Velez is an Associate Professor of Medicine at Ochsner Clinical School / The University of Queensland and Interim Chair of the Department of Nephrology at Ochsner Clinic Foundation in New Orleans, LA. His translational research efforts involve diagnostic strategies to better phenotype individuals with AKI and cirrhosis to improve implementation of therapeutic interventions. Follow him @VelezNephHepato.
Writer: Bill Whittier @TWhittier_RUSH
Bill Whittier is an Associate Professor practicing Nephrology at Rush University Medical Center in Chicago, IL. His main research area involves the adequacy and safety of the renal biopsy. Other research publications are in the realm of lupus nephritis, glomerulonephritis, and diabetic nephropathy. He has also written on topics of acid-base disorders and hyponatremia.
Competitors for the Hepatorenal Region
Bile Casts: Pathologic vs Bile Casts: Incidental
Terlipressin vs Norepinephrine

Copyright: Magic mine / Shutterstock
Patients with liver failure are sick. Patients with acute kidney injury (AKI) are sick. Put the two together and you have a fine mess. For the first time NephMadness is taking these two conditions and putting them into their own region. The management of hepatorenal syndrome (HRS) has improved over the last 20 years with the use of intravenous albumin for resuscitation, the aggressive prevention and treatment of spontaneous bacterial peritonitis, and the adoption of therapies that treat splanchnic vasodilation to improve renal hemodynamics and reverse the primary pathology. While considering the field of HRS, selection committee member, Juan Carlos Velez (with a twitter handle like @VelezNephHepato, could we have done any better?), selected two important matchups:
- Bile acids. Increased bilirubin is reliably found in liver failure. Bile acid forms casts in the kidney. These are often found on autopsy after a patient dies with kidney failure. This question devolves into simply whether these bile acid casts are an incidental finding or pathologic and the cause of the AKI.
- Is the improvement in HRS outcomes found with terlipressin and other strategies to increase splanchnic vasoconstriction working as advertised or is the critical change merely an increase in the systemic blood pressure? This match-up pits terlipressin representing splanchnic vasoconstriction against norepinephrine representing increased systemic pressure.
Bile Acids: Pathologic
The idea that bile pigments might be toxic began in 1899 when Quincke noted bile pigments staining the glomeruli in autopsies of patients with acute onset jaundice. It doesn’t appear that Quincke published this work, but it was reported in Hessler’s 1922 account of the bilirubin content of urine and that it correlated with the degree of jaundice. In 1937, Elsom showed that jaundice was associated with decreased kidney function and that improvement in jaundice was associated with improved in kidney function. The idea that bilirubin was responsible for decreased renal function in cirrhosis, a condition called cholemic nephropathy, did not gain much traction and was rarely reported in the literature until 2013 when van Slambrouck et al reported a pathology series from 44 jaundiced patients, 41 autopsies, and 3 kidney biopsies. In 24 of these patients, they found bile casts. The presence of casts was correlated with higher bilirubin levels and a trend towards higher creatinine.

Visual Abstract by @kidney_boy on van Slambrouck et al
Of course the best part of the article was finding out that formalin-fixed kidneys turn green as the bilirubin is converted to biliverdin.
Before formalin. Image courtesy of Anthony Chang @ChangUCanSpare

After formalin. Image courtesy of Anthony Chang @ChangUCanSpare
If bile casts are pathologic, how would they cause kidney damage? A study of 35 patients with obstructive jaundice found evidence of proximal tubule dysfunction with increased urine uric acid (uricuria), glucosuria, and phosphaturia. The tubular damage was reversible following correction of the jaundice lending weight to Slambruck’s suggestion that bile casts may be directly nephrotoxic. Similar to myeloma and myoglobin casts, bile acid casts could cause tubular obstruction. Bile acids are poorly soluble in water and this may contribute to their formation of casts in the acidic environment of the kidneys.
Fickert et al created a mouse model of cholestatic jaundice via common bile duct ligations in mice. They were able to show liver injury, hepatic fibrosis. They also showed increased BUN, dilated renal tubules, interstitial nephritis, and fibrosis at 8 weeks. The experiment was run with Farnesoid X receptor knockout mice. Farnesoid X receptor is important in the regulation of bile acid production and results in mice forming more hydrophillic bile casts. Farnesoid X receptor knockout mice did not develop kidney fibrosis following common bile duct ligation. Similar finding was found when switching the mice to a diet rich in hydrophilic norursodeoxycholic acid, which also results in more hydrophilic bile acids.

Visual Abstract by @Errantnephron on Fickert et al
What may be the most important and lasting impact of Slambrouck’s study was changing the name from cholemic nephropathy to bile cast nephropathy. The new name caught the imagination of nephrologists and initiated a steady drumbeat of case reports using the new name.
Patel et al reported a case of presumed bile acid nephropathy. The physicians discussed that bile acid nephropathy and AKI occurred when the bilirubin rose above 10 mg/dL. The patient became dialysis-dependent. The physicians initiated plasmapheresis to lower the bilirubin (bilirubin is highly protein-bound and is not appreciably affected by conventional dialysis). When the bilirubin fell below 10 mg/dl, kidney function recovered and the patient stopped dialysis (but continued plasmapheresis). The patient then lost her tunneled catheter for plasmapheresis, and the bilirubin rose, the renal function deteriorated, and the patient became dialysis-dependent once again. The patient ultimately received a liver transplant, bilirubin fell to normal levels, and kidney function returned to normal.
Fisler et al reported a case of anabolic steroid induced liver failure followed by AKI. A kidney biopsy revealed dilatation and necrosis of the renal tubules, as well as intraluminal pigmented casts consistent with bilirubin casts. nephropathy.
Aniort et al described a case of bile cast nephropathy following an episode of obstructive cholestasis due to gallstones. Liver biopsy revealed cholestatic hepatitis but no cirrhosis or ascites. His creatinine went to 5.3 mg/dL while the urine sodium was 95 mmol/L, making hepatorenal physiology unlikely (consistent with the lack of cirrhosis and ascites). A pair of ERCPs cleared the stones and obstruction. Three months later, renal function returned to normal.
Bile cast nephropathy is an old player showing new life in acute kidney disease with liver dysfunction. The data on this condition is thin and we are in need of consensus definitions of what determines a case, but when the bilirubin is high and the kidneys aren’t working this entity should be considered. Given its up and coming status, “Bile Acids are Nephrotoxic” should advance in NephMadness.
Bile Casts: Incidental
One of the first lessons in basketball is to keep your eye on the ball. Don’t be distracted by associations that are not driving the pathology. The whole field of nephrology was electrified by the van Slambrouck et al case series that really put this disease back on the map, so it is valuable to take a look at the reaction it generated at the time.
In a letter to the authors, Heyman et al point out that van Slambrouck’s landmark paper is merely observational and cannot prove causality. They theorize that the low GFR may prevent normal wash out of casts resulting in the pathologic findings of the study. They also point to literature showing an antioxidant effect of bilirubin and evidence that it can be nephroprotective in some situations.
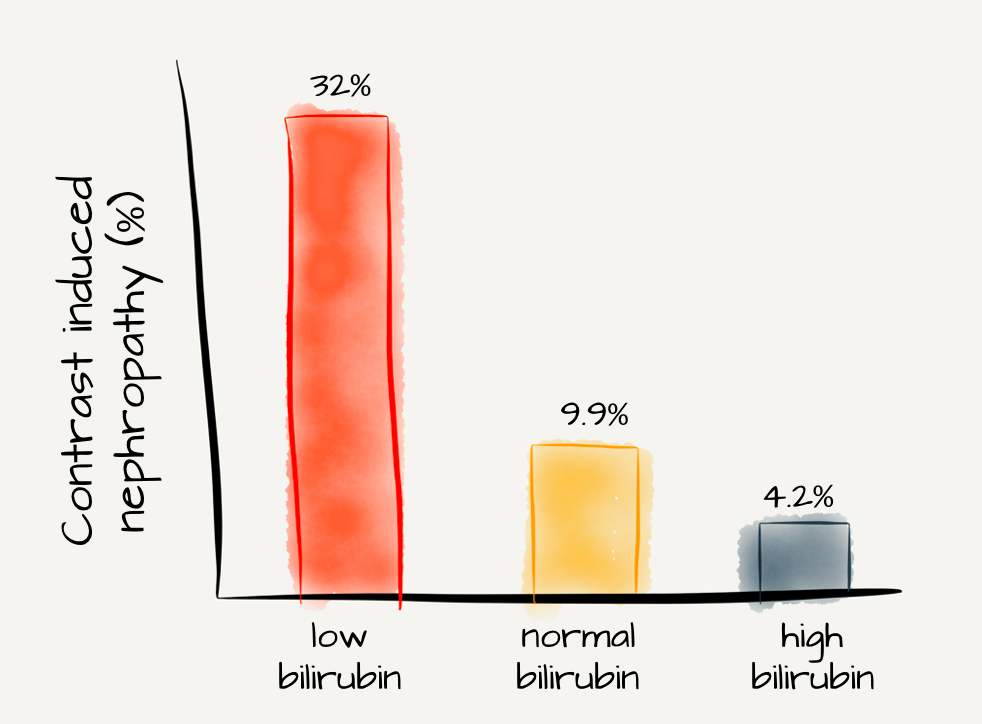
Adapted from Figure 1 Huang et al, PLoS One
As crazy as it sounds, that is not the only study that has found bilirubin to be nephroprotective. Oh et al found that bilirubin protected proximal tubule cells from cyclosporine nephrotoxicity. In another win for team bilirubin, Leung et al used glycerine to induce AKI; common bile duct ligation reduced the kidney injury and mortality. How about Barabas’ study of Gunn rats that showed increased bilirubin to be nephroprotective against cisplatin nephrotoxicity? And probably most convincing was Park et al’s cohort study of over 1,000 patients. They found slower progression of CKD in patients with slightly elevated total bilirubin (0.8 to 1.2). Similarly, Deetman et al found elevated bilirubin levels were protective against graft failure in kidney transplant patients.

Image courtesy of Anthony Chang @ChangUCanSpare
One of the problems with the diagnosis is that bile casts are found in other conditions. Nayak et al did an autopsy study of 125 patients who died with a clinical diagnosis of HRS. Bile casts were found in 44% of patients. Does this study suggest that our clinical diagnosis of HRS is wrong nearly half the time or are bile casts just an incidental finding in kidney biopsies and not really causing harm? The crux of this issue is that the finding of bile casts on biopsy does not mean the AKI is due to these casts. This is an exceedingly important issue to clarify as we move toward evidence-based interventions for HRS. If nearly half of these cases are caused by what had been previously thought to be an innocuous biopsy finding, then almost all HRS trials will be helplessly underpowered.
Regardless of the speculation on the pathogenesis of bile cast nephropathy, the cause of AKI in patients with liver failure and/or hyperbilirubinemia is diverse, and should not always be blamed on HRS. Nevertheless, adjudicating a diagnosis of bile cast nephropathy to any AKI with high bilirubin does not seem to be supported by any solid evidence of causality. In a very important contribution from Alsaad and Wadei, none of 88 patients with cirrhosis who underwent kidney biopsy as part of evaluation for dual organ transplantation were found to have bile cast nephropathy. Zero. Of note, 12 had ATN and 10 had normal kidneys (i.e., biopsy-proven HRS). Thus, it seems unlikely that a real entity could be missed in such large cohort. Therefore, bile casts as incidental findings should advance in this round of NephMadness.

Visual Abstract by @whatsthegfr on Alsaad and Wadei
Check out this podcast episode of The Curbsiders featuring @VelezNephHepato @TWhittier_RUSH @kidney_boy:
#145 NephMadness: Hepatorenal Syndrome vs AKI
Terlipressin
In 1956, Hecker and Sherlock published a post-mortem series of 9 patients with cirrhosis who all developed kidney failure and died. The kidneys showed normal histology and the diagnosis of HRS (though they did not use that term in their paper) was born.

Excerpt from Hecker and Sherlock, The Lancet
The pathophysiology of HRS is critical to understanding the role terlipressin plays in its treatment. Patients with cirrhosis have portal hypertension which leads to splanchnic vasodilation and a reduction in systemic vascular resistance. In response to this drop in SVR, there is activation of the adrenergic and renin-angiotensin-aldosterone systems to maintain arterial blood pressure. As cirrhosis and portal hypertension advances, hemodynamic stability is more and more dependent on vasoconstriction of the extra-splanchnic vascular beds. This intense vasoconstriction leads to decreased renal perfusion and GFR. Cirrhosis can depress cardiac activity further decreasing kidney perfusion and GFR.
Because the initiating event in the cascading hemodynamic failure of HRS is splanchnic vasodilation, a logical therapeutic target is reversing the splanchnic vasodilation. The drug most studied for this is terlipressin. Terlipressin is a prohormone of lysine-vasopressin. Following intravenous administration, 3 glycyl residues are metabolized by endothelial peptidases producing active lysine-vasopressin. Being a product that requires endogenous activation prolongs the half-life, so terlipressin can be given in divided doses (unlike vasopressin, which must be given by continuous IV drip). Terlipressin binds both V1 and V2 receptors.

Copyright: Numstocker / Shutterstock
Within 30 minutes, terlipressin decreases hepatic resistance and renal arterial resistance (reversing the portal hypertension and renal vasoconstriction). This correlates with a drop in plasma renin activity and decreased activation of the sympathetic nervous system.
But the hemodynamic changes don’t mean a thing if patients don’t get better. HRS has a case fatality rate of 47%. In 2018, Wang et al did a systematic review and meta-analysis of the randomized controlled trials on terlipressin. They limited their analysis to trials of terlipressin alone or with albumin versus placebo, albumin, or other vasoconstrictors. The primary outcomes of analysis were:
- HRS reverse (creatinine < 1.5 mg/dl)
- Renal function change (50% drop in creatinine, but still greater than 1.5 mg/dL)
- Mortality
They found 18 RCTs representing 1,011 patients (509 with terlipressin and 502 controls). Terlipressin doses ranged from 1.5 to 12 mg a day. The 18 studies breakdown like this:
| Terlipressin versus placebo | 7 |
| Terlipressin versus norepinephrine | 8 |
| Terlipressin versus octreotide | 1 |
| Terlipressin versus dopamine | 1 |
| Terlipressin versus octreotide and midodrine | 1 |
Terlipressin was much better than placebo for all outcomes including mortality. Compared to norepinephrine it was equivalent for all outcomes, except it had a higher rate of adverse events (though the authors were skeptical of this finding due to the way adverse events were reported). Terlipressin was superior than octreotide and also terlipressin plus albumin was superior to octreotide, midodrine, and albumin. There was no difference between terlipressin and dopamine in the one study that looked at it; however, there were very few outcomes examined in that study (no measure of renal function was provided so no comment can be made for dopamine’s relative ability to reverse or improve renal function). Overall, the meta-analysis found HRS reversal in 42% of the patients on terlipressin versus 26% in the controls. Type 2 HRS and a lower creatinine made reversal more likely. From the conclusion, Wang et al points out that despite methodological differences from other systematic reviews:
…our study had the same conclusion with the others. All the previous meta-analysis involving HRS reverse found that terlipressin increased the number of patients with reversal of HRS.
But despite the homogeneity of these meta-analysis, terlipressin is still not available in the US. Wang et al noted that only two of the RCTs they looked at had an N > 100. Currently, Mallinckrodt is conducting a quadruple blind (participants, providers, investigators, and outcome assessors) randomized, placebo-controlled trial in the US and Canada. It is scheduled to be completed in November of 2019. Look for The CONFIRM Study to confirm that terlipressin was the appropriate winner of NephMadness sometime in 2020.
And if you are still thinking about going the other way and voting for norepinephrine, take a look at this RCT from August of 2018 between terlipressin and norepinephrine with 120 patients. Terlipressin reversed HRS in 40% of patients versus 17% with norepinephrine, P value 0.004. It also significantly reduced renal replacement therapy and improved survival.
Norepinephrine
Part of the origin story of terlipressin is that it is a splanchnic vasoconstrictor, and its ability to reverse deadly HRS is a direct result of this splanchnic vasoconstriction. But let’s look at the tape carefully. Is that splanchnic vasoconstriction really how this plays out? The hepatorenal cocktail is an albumin infusion combined with either terlipressin (where available) or midodrine plus octreotide if unavailable (America).

Copyright: sfam_photo / Shutterstock
The goal is to use preferential vasoconstriction of the splanchnic circulation to shunt more renal blood flow to improve GFR. The benefit of albumin plus terlipressin was initially shown in a series of nine consecutive patients with HRS where 7 of the 9 had HRS reversal (creatinine below 1.5 mg/dL). Of note in that study is that mean arterial pressure (MAP) went from 68 to 80 mm Hg. Terlipressin alone versus terlipressin plus albumin was studied further in 21 patients, and the combination was found to be superior at reversing HRS (25% vs. 77%). In the terlipressin alone group, the MAP decreased from 64 to 60 mm Hg, but in the combination group, the MAP increased from 70 to 79 mm Hg.

Adapted from Figure 2 in Ortega et al, Hepatology
Further evidence of the importance of raising blood pressure came when examining the studies using octreotide and midodrine. One of the first studies from 1999 paired octreotide and midodrine against low dose dopamine in 13 patients. The combination improved MAP by 16 mm Hg and improved the HRS in 60% compared to no change in blood pressure or improvement in HRS in the dopamine group. Sixteen years later, octreotide and midodrine squared off against terlipressin, and terlipressin advanced as the winner. However, the positive responders in each group demonstrated increases in MAP.
We are certainly not the first people to notice this; selection committee member, and sponsor of this region, Dr. Juan Carlos Velez, looked at 21 studies on 501 patients receiving vasoconstrictor therapy and found a tight association with improvement in MAP and serum creatinine.

Correlation between increase in mean arterial pressure (MAP) and change in kidney function. Diameter of each data point reflects relative sample size of cohort. Figure 2 from Velez and Nietert, AJKD, © National Kidney Foundation.
Most of the data came from terlipressin, but the analysis included studies of midodrine and octreotide, norepinephrine, and albumin. The point of the study is that the agent isn’t important, it’s the change in blood pressure. The strategy is no longer limited to splanchnic vasoconstriction but invites all different ways to improve blood pressure. Even though we are unable to get the terlipressin in the US, we do have access to some fairly effective vasopressors. And the one representing this arm of the match-up is among the most effective, norepinephrine. In fact, if you look at the very first description of HRS by Hecker and Sherlock in 1956, this agent was used with some apparent success:

Excerpt from Hecker and Sherlock, The Lancet
So what does the data look like in the post-1956 world? The earliest data comes from 2002: Duvoux et al gave twelve consecutive patients with HRS norepinephrine, albumin, and furosemide. The renal function improved and HRS reversed in 10 of the 12, and MAP rose from 65 mmHg at baseline to 73 mmHg. In 2013, Badawy et al published a RCT of patients that had failed octreotide and midodrine. In this cohort, there was no difference between change in renal function (which did improve) and survival at 30 days. The notable difference? Norepinephrine was one quarter the cost of terlipressin. Saif et al found similar results in this 60 person RCT from India and Italy showing no difference between norepinephrine and terlipressin. If you are a fan of systematic reviews, check out Nassar Junior et al’s analysis: “Norepinephrine seems to be an attractive alternative to terlipressin in the treatment of HRS and is associated with less adverse events”.
The above mentioned2018 paper by Arora et al that reported superiority of terlipressin continuous infusion over noradrenaline in the treatment of acute kidney injury has some major concerns. First, previous data indicate that continuous infusion of terlipressin does not lead to more HRS reversal than the standard bolus dosing of terlipressin. Secondly, the authors report an unprecedented 80% rate of renal replacement therapy in the norepinephrine group. Thirdly, while subjects enrolled in a placebo-controlled terlipressin RCT in the US had a baseline serum bilirubin of 11 – 12 mg/dL and a HRS reversal rate of 13 – 20%, Arora et al reported a 40% HRS reversal rate in subjects receiving terlipressin with bilirubins around 22 mg/dL. This misalignment is very hard to reconcile and calls for critical analysis. The open-label nature of the study might be a factor here. Caution should be used before accepting this paper as a practice changer.
Today, we live in a world where, at least in the US, terlipressin is not available, and in other parts of the world it’s available but at high costs. It is possible that sometime in 2020, terlipressin will be approved by the FDA, but stop your hand-wringing and focus on the MAP. You can raise the MAP with norepinephrine to get all of the advantages of terlipressin with neither the cost or adverse effects.
– Executive Team Member for this region: Joel Topf, AJKD Social Media Advisory Board member. Follow him @kidney_boy.
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC point for reading this region.
- Register/log in to the NKF’s Professional Education Resource Center (PERC).
- Review the activity and accreditation information.
- Click “Continue” and review Course Instructions.
- Complete Post-Test. Please note: By selecting “Yes” to the participation questions for each region, the corresponding Post-Test questions will appear. Click “Save Draft” to save your responses and finish later. When you are ready to submit your answers, click “Preview” to review all responses, then click “Submit.”
- Click “Next” to complete the Evaluation form, then click“Submit.”
- Claim 1.0 CME credit and 1.0 MOC point per region (up to 8.0 total for 8 regions of NephMadness).
- Print your certificate.
- Review the Post-Test answers and rationale.
The CME and MOC activity will expire on June 15th, 2019.

Leave a Reply