
Eculizumab – Can It Be Used for MPGN and C3GN?
Membranoproliferative glomerulonephritis (MPGN) is a pattern of glomerular injury on light microscopy that is characterized by mesangial expansion with hypercellularity, endocapillary proliferation, and double-contour formation along the glomerular capillary walls. MPGN has traditionally been classified as types I, II, and, III based upon electron microscopic findings.

Traditional classification of membranoproliferative glomerulonephritis (MPGN)
Substantial progress in our understanding of the relationship between abnormal regulation of the complement system and glomerular inflammation has led to a new classification and a recently defined entity called C3 glomerulopathy (C3G) (Figure 2).
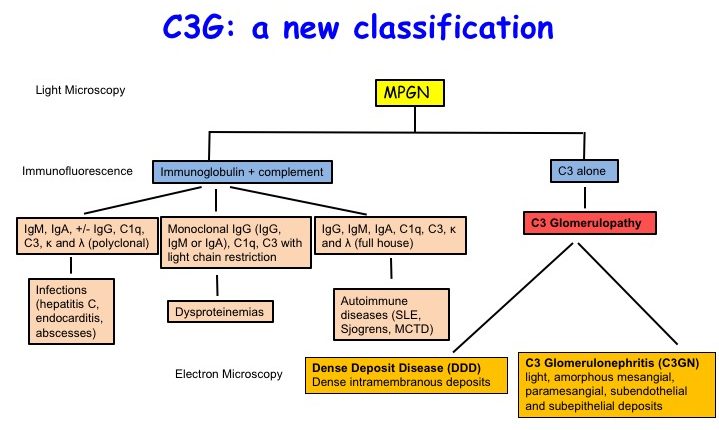
C3 glomerulopathy (C3G), a new classification scheme. Adapted from Figure 2 from Daccueil et al, AJKD © National Kidney Foundation.
C3G encompasses a group of kidney diseases driven by uncontrolled activation of the complement cascade that leads to C3 deposition within the glomerulus. It consists of dense deposit disease (DDD) and C3 glomerulonephritis (C3GN). DDD is a specific form of C3G that is classified by the presence of dense osmiophilic intramembranous deposits seen on electron microscopy. C3GN is a form of C3G in which deposits on electron microscopy may be light and located in the mesangial, subendothelial, or subepithelial regions. This new classification based on the pathogenetic process helps to direct the clinical evaluation and to provide disease-specific treatments.
Important points to note:
- A diagnosis of C3G requires a renal biopsy
- A diagnosis of C3G can only be made on immunofluorescence (IF).
- Criterion on IF: Presence of dominant C3 staining at least 2 orders of magnitude greater than any other immunoreactant (i.e IgG, IgA, IgM, or C1q)
- EM should be conducted in all cases to distinguish DDD and C3GN
- Once C3G has been diagnosed on IF, light microscopy can then help identify diverse patterns of glomerular injury that may include MPGN and further detect additional features such as crescentic disease, interstitial fibrosis, and tubular atrophy
The primary process that leads to glomerular C3 deposition in C3G is the dysregulation of the alternative pathway (AP) of complement activation. Complement activation is designed to eliminate the pathogen and clear debris. However, if misdirected or dysregulated, it can be similarly damaging to self. Therefore, strict regulation of the complement system is required to prevent collateral damage to host cells and tissues.
The regulatory proteins that protect against undesirable AP activation include two in plasma, factor H (FH) and factor I (FI), and three membrane proteins, namely, membrane cofactor protein (MCP; CD46), decay-accelerating factor (DAF; CD55), and complement receptor 1 (CR1; CD35). Inherited defects in these complement genes and acquired autoantibodies against a regulatory protein predispose to several diseases because of inadequate control of complement activation.
Notably, evidence of uncontrolled complement activation can also be found in cases of MPGN that do not meet diagnostic criteria for C3G. These are termed immune-complex MPGN (IC-MPGN) and result from activation of the classical pathway of complement by antigen-antibody complexes. Therefore, uncontrolled complement activation can lead to both C3G and immune-complex MPGN.

Complement activation may occur through the classical, lectin, and alternative pathways. The classical pathway is primarily activated when C1q binds to antibodies complexed with antigens. The lectin pathway is activated when mannose-binding lectin (MBL) binds to mannose-containing proteins or carbohydrates on the surface of a pathogen. The alternative pathway is activated by the direct binding of C3 to pathogens or “altered self”. All three pathways converge to form C3 convertase which further converts C3 to C3a and C3b. C3b in the presence of Factors B and D generates even more convertase resulting in a potent amplification loop (represented by the dashed red line). C3b opsonizes bacteria that phagocytes recognize that enables engulfment and destruction. Addition of C3b to a C3 convertase generates a C5 convertase that cleaves the C5 protein into C5a (a potent anaphylatoxin) and C5b which activates the terminal complement complex pathway resulting in the formation of the membrane attack complex (MAC) on cell surface thereby resulting in cell lysis.
The genetic drivers of disease include rare or novel variants in complement genes, identified in ~ 30%-40% of patients, while the acquired drivers include C3 nephritic factors (C3Nefs), FH autoantibodies, and presence of monoclonal gammopathy (see the NephMadness 2019 Complement Region to learn more). Of the acquired factors, the most common and the best studied are C3Nefs reported in up to 80% of patients with DDD and 50% of patients with C3GN. C3Nefs are autoantibodies that bind to and stabilize the AP C3 convertase (C3bBb) by preventing its inactivation by FH. This leads to a prolonged C3 convertase activity resulting in continued conversion of C3 to C3b, which in turn consumes the C3. These autoantibodies may also stabilize the C5 convertase, leading to massive activation of the terminal complement pathway and generation of very high levels of sC5b-9.
Unlike in aHUS, the role of complement inhibition by eculizumab in C3G and MPGN is not clear and data are conflicting. Nonetheless, eculizumab use has been reported in a number of cases of C3G and MPGN. Current reports suggest a more beneficial role for eculizumab in cases with prominent terminal pathway activity.
In a recently published report in AJKD, Ruggenenti et al evaluated the effect of eculizumab in 10 patients (6 with biopsy-proven MPGN and 4 with C3G) identified between March 2014 and January 2015 from 6 centers. Five patients were children or adolescents. This was a single-arm, prospective, open-label study organized into two 48-week periods of treatment with eculizumab, divided by a 12-week washout period (where eculizumab was withdrawn).

Eculizumab treatment plan
Inclusion Criteria:
- Creatinine clearance > 20 ml/min/1.73 m2
- Proteinuria > 3.5 g/day in adults; >40 mg/m2/h in children
- Serum C3 < 80 mg/dl
- Plasma sC5b-9 levels > 1000 ng/ml on at least 2 consecutive measurements performed 10 weeks apart
Exclusion Criteria:
- Age >75 years
- Secondary MPGN
- Severe chronic kidney histologic changes (>50% glomeruli globally sclerotic and/ or >50% renal cortex with interstitial fibrosis and atrophy)
- Use of steroid or immunosuppressive therapy in the past 6 months
- Inability to understand the risks and benefits of the study
- Pregnant or lactating women
Primary Outcome: 24-hour proteinuria reduction at weeks 24 and 48 weeks of the first treatment period as compared to baseline
Secondary Outcomes:
- Change in GFR (as measured by the iohexol plasma clearance technique and estimated using the CKD-EPI equation in patients older than 18 years; Schwartz formula in younger patients) – measured at weeks 1, 12, and 24 of the first treatment period; 12 weeks after treatment withdrawal; and at weeks 24 and 48 of the second treatment period
- Albumin and IgG fractional clearances measured at the same time points as GFR above
- Complete remission of nephrotic syndrome (protein excretion < 0.3 g/day) measured at weeks 1, 12, 36, and 48 weeks of both treatment periods
- Partial remission of nephrotic syndrome (proteinuria excretion <3.5 g/day with >50% reduction from baseline) measured at weeks 1, 12, 36, and 48 weeks of both treatment periods
- Plasma sC5b-9 levels measured at weeks 1, 12, 36, and 48 weeks of both treatment periods
- Serum C3 levels measured at weeks 1, 12, 36, and 48 weeks of both treatment periods

Changes over time in (first panel from the top) sC5b-9 (serum complement membrane attack complex) plasma levels; (second panel) C3 serum levels, (third panel) serum albumin concentration, (fourth panel) 24-hour proteinuria, and (fifth panel) measured (mGFR) and estimated (eGFR) glomerular filtration rate (GFR) during the first (period a) and the second 48-week treatment period (period b), which were separated by a 12-week washout period. Figure 2 from Ruggenenti et al, AJKD © National Kidney Foundation.
Results:
- Proteinuria reduction at weeks 24 and 48 (primary outcome) as compared to baseline achieved statistical significance in 3 of 10 patients (2 with IC-MPGN and 1 with C3G). Among these 3 responders, proteinuria increased back to baseline during the withdrawal period. Changes in albuminuria and IgG fractional clearances paralleled the change in proteinuria.
- Two of these patients achieved partial remission at the end of both treatment periods and the third patient achieved partial remission at the end of the second treatment period.
- All 3 patients achieving partial remission were C3Nef negative.
- In the remaining 7 patients, eculizumab had no effect on proteinuria reduction and 5 of the 7 were positive for C3 nephritic factors.
- One patient progressed to ESRD during the second treatment period.
- Mean measured GFR transiently increased during the first treatment period, but declined thereafter during the washout and second treatment period.
- Plasma sC5b-9 levels promptly and persistently normalized during the first treatment period, recovered to baseline at the end of the washout period, and normalized again during the second treatment period.
- Serum C3 levels were persistently reduced throughout the study period but transiently increased during the washout period.

Changes over time in (first panel from the top) sC5b-9 (serum complement membrane attack complex) plasma levels, (second panel) C3 serum levels, (third panel) 24-hour urinary protein excretion, and (fourth panel) measured glomerular filtration rate (mGFR) during the first (period a) and the second 48-week eculizumab treatment period (period b), which were separated by a 12-week washout period, in 3 patients achieving partial remission at study end (continuous line) and 7 patients who did not achieve this end point (dashed line). Figure 3 from Ruggenenti et al, AJKD © National Kidney Foundation.
Safety:
Acute reactions occurred in 11.6% infusions. These included chest pain, nausea, headache with blurred vision, transient temporal hemianopsia, and hypotension. One patient was hospitalized for pneumococcal pneumonia but recovered fully with treatment.
Discussion:
This was a single-arm exploratory study conducted to investigate the effect of eculizumab in patients with MPGN/C3G. Proteinuria decreased in only 3 patients despite solid evidence that the membrane attack complex was blocked leading to normalization of sC5b-9 levels in all patients. This likely indicates that disease activity in C3G is only partially mediated by the terminal complement pathway and that other upstream mechanisms play a key role in the pathogenesis of this . Importantly, 5 of the 7 non-responders were positive for the C3Nef autoantibodies, further suggesting that eculizumab has no effect on these antibodies, which are likely better treated with rituximab or plasmapheresis.
Mutations in complement proteins (D1625H in C3 and R78G in Factor H) were identified in two patients with C3G. Although the authors report that these variants were classified as mutations based on an allele frequency < 0.001 along with functional data supporting pathogenicity of the variants, the actual data were not reported.
To date, the information of the efficacy of eculizumab in MPGN/C3GN had only been in the form of case reports. Ruggenenti et al are to be congratulated in putting together this trial. However, it was disappointing that treatment with eculizumab was of minimal efficacy with only a modest effect on GFR. In patients with C3G, other treatments need to be devised that block earlier in the cascade (Factor D, Factor B, or C3). Trials are currently ongoing for these novel targets.
– Post prepared by Anuja Java, AJKDBlog Guest Contributor. Follow her @anuja_java.
To view Ruggenenti et al (subscription required), please visit AJKD.org.
Title: C5 Convertase Blockade in Membranoproliferative Glomerulonephritis: A Single-Arm Clinical Trial
Authors: P. Ruggenenti, E. Daina, A. Gennarini, C. Carrara, S. Gamba, M. Noris, N. Rubis, F. Peraro, F. Gaspari, A. Pasini, A. Rigotti, R.M. Lerchner, D. Santoro, A. Pisani, A. Pasi, G. Remuzzi, on behalf of the EAGLE Study Group
DOI: 10.1053/j.ajkd.2018.12.046
thank you for the information.