
Autosomal Dominant Tubulointerstitial Kidney Disease – What are MUC and MOD?
Autosomal dominant tubulointerstitial kidney disease (ADTKD) was first described in 1944 and has many names – medullary cystic kidney disease type 1 and type 2 or familial juvenile hyperuricemic nephropathy type 1 and type 2. Reports in the early 1980s suggested that juvenile nephronophthisis and medullary cystic kidney disease are the same disease. The names were a hint that this disease group was inherited and associated with medullary cysts and gout. As one can imagine, these diagnoses based on clinical features, imaging, and uric acid levels were confusing. Today, we know that the clinical features of this group of kidney diseases, now defined genetically, are very variable between and within families. The unifying clinical features of autosomal dominant tubulointerstitial kidney disease are:
1) autosomal dominant inheritance,
2) slowly progressive chronic kidney disease, and
3) bland urine with no or minimal proteinuria.
Medullary cysts or gout are NOT required for a diagnosis (in fact, medullary cysts are rare).

Samples of kidney biopsy of patients with ADTKD: patient with (A) ADTKD-MUC1 and (B) ADTKD-UMOD. Both periodic acid–Schiff staining (original magnification, ×20) show extensive interstitial fibrosis with glomerulosclerosis and tubular atrophy at the deep cortex. Mild mononuclear interstitial infiltrate is also seen. Figure 1 from Ayasreh et al, AJKD © National Kidney Foundation.
The first investigation into the genetic cause of this disease occurred in 1998. This study of a family with medullary cystic kidney disease type 1 mapped to the gene locus 1q21. The gene turned out to be MUC1 encoding the mucin-1 protein. Other genes have now been identified in this disease spectrum: UMOD, REN, and HNF1B. ADTKD is currently subdivided according to the gene mutated, which include all of the above. UMOD and MUC1 are the most common genes mutated, while ADTKD-HNF1B mutations are very rare.
Overall, ADTKD is a rare disease with ADTKD-UMOD occurring in less than 2000 families worldwide, ADTKD-MUC1 in less than 1000 families, and ADTKD-REN in less than 200 families. Some clinical features are associated with ADTKD subtypes. Hyperuricemia can present at an early age in ADTKD-UMOD and is thought to be due to increased proximal reabsorption of urate. This feature cannot be relied upon for diagnosis as it is variably present and hyperuricemia is also common in CKD of any type. Hyperuricemia can also occur in ADTKD-REN and is thought to occur due to increased proximal urate reabsorption as a result of low blood pressure, another clinical feature of ADTKD-REN.
The recent AJKD publication by Ayasreh et al describes the clinical and genetic findings in a Spanish cohort of patients with ADTKD. This is a large cohort for a rare disease such as ADTKD, which makes their findings important. 131 individuals from 56 families were investigated for the presence of UMOD and MUC1 mutations. They found mutations in 25 families and the authors performed in-depth phenotyping analysis in these individuals. Nine families (41 individuals) had a UMOD variant they described as likely pathogenic and 16 families (90 individuals) had a MUC1 mutation. All of the MUC1 mutations were cytosine duplications in a variable number of tandem repeats (VNTR) as described in other ADTKD cohorts. It is this VNTR feature that makes it difficult to sequence this MUC1 variant using standard sequencing methods.
Ayasreh et al then compared clinical features between the two groups, ADTKD-UMOD and ADTKD-MUC1. Earlier age at diagnosis occurred with ADTKD-UMOD (32 vs 40). Polyuria and polydipsia were not significantly different between groups but occurred more frequently with UMOD. Hyperuricemia was significantly more common with UMOD than MUC1 (87% vs 54%, respectively), although gout was not statistically significant between the groups. These findings are somewhat consistent with the old name of familial juvenile hyperuricemic nephropathy for the UMOD group. The rate of GFR decline was not different between groups, with the age of ESRD onset being 51 years (95% confidence interval [CI], 47-57).

Clinical Features for ADTKD-UMOD and ADTKD-MUC1 Groups. Mean ± standard deviation are presented for quantitative variables, and n/N (percent), for categorical variables. Table 1 from Ayasreh et al, AJKD © National Kidney Foundation.
Ultrasound findings of cysts occurred in the minority of patients (13% for UMOD vs 26% for MUC1).
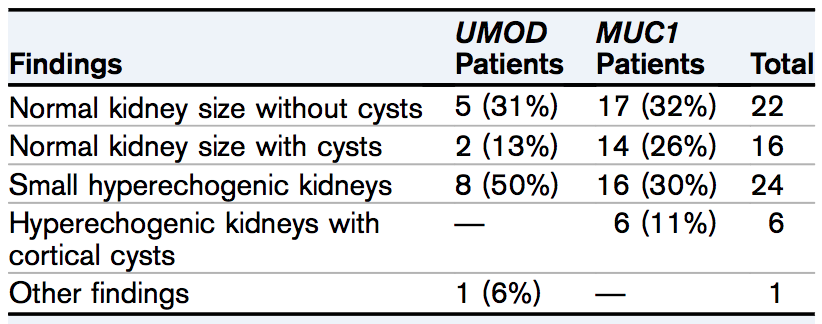
Renal Ultrasound Findings for ADTKD-UMOD and ADTKD-MUC1 Groups. Table 2 from Ayasreh et al, AJKD © National Kidney Foundation.
In the cohort of 56 families, there were no REN or HNF1B mutations found, and gene mutations were identified for only 45% of the families. There may be other mutations in the MUC1 gene that can cause this disease but have not yet been identified due to the technical difficulty of sequencing this gene. It is presumed that some of the families in this cohort may have undiscovered MUC1 mutations. Other families might have UMOD gene variants that were defined by this study as not disease-causing (Ayasreh et al used bioinformatics to determine the likelihood that missense variants are disease-causing). The high degree of variability within this disease group is likely explained by modifier genes yet to be described. For example, some undiagnosed families in this cohort may have disease due to genetic epistaxis between a modifier gene locus and a UMOD mutation that on its own looks (bioinformatically) benign.
Ayasreh et al should be applauded for characterizing the genetic and clinical features of this group of rare but interesting kidney diseases. Hopefully, with increased awareness of this disease, more causative genes will be identified which may lead to new therapies for these patients.
– Post prepared by Andrew Malone, AJKDBlog Contributor. Follow him @AndrewFMalone.
To view Ayasreh et al (subscription required), please visit AJKD.org.
Title: Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1
Authors: N. Ayasreh, G. Bullich, R. Miquel, M. Furlano, P. Ruiz, L. Lorente, O. Valero, M.A. García-González, N. Arhda, I. Garin, V. Martínez, V. Pérez-Gómez, X. Fulladosa, D. Arroyo, A. Martínez-Vea, M. Espinosa, J. Ballarín, E. Ars, and R. Torra
DOI: 10.1053/j.ajkd.2018.03.019
Leave a Reply