
#NephMadness 2022: Cardiorenal Region
Submit your picks! | NephMadness 2022 | #NephMadness | #CardioRenal
Selection Committee Member: Nisha Bansal @NishaKidneyDoc
Nisha Bansal is an Associate Professor and the Arthur Stach Family Endowed Professor in the Division of Nephrology at the University of Washington (UW). She is also an investigator in the Kidney Research Institute, the Associate Fellowship Program Director and the Director of the Kidney-Heart Service at UW. She is an expert in the pathophysiology, diagnosis, and treatment of hypertension and cardiovascular disease in patients with chronic kidney disease and those treated with dialysis.
Selection Committee Member: Sadiya Khan @HeartDocSadiya
Sadiya Khan is a cardiologist at Northwestern Medicine and an Assistant Professor in the departments of Medicine and Preventive Medicine at Northwestern University Feinberg School of Medicine. Her research focuses on epidemiology, risk prediction, and mechanisms of heart failure across the life course.
Writer: Caitlyn Vlasschaert @DrFlashHeart
Caitlyn Vlasschaert is an internal medicine resident and PhD candidate at Queen’s University in Canada. Her research is focused on somatic myeloid cell mutations in kidney disease. She is a graduate of the 2020 class of the Nephrology Social Media Collective (NSMC) Internship and now sits on its executive committee.
Competitors for the Cardiorenal Region
Diuretic Resistance Mechanisms vs Diuretic Resistance Treatments
Cardiac Biomarkers in CKD vs Kidney Biomarkers in CHF

Copyright: Sri J / Shutterstock
Diuretic Resistance Mechanisms vs Diuretic Resistance Treatments
Achieving adequate diuresis in patients with decompensated heart failure and other edematous states has been a longstanding challenge in medicine, and can sometimes feel like sinking a shot from well beyond the three-point line — while surrounded by defenders. This challenge dates back to ancient Greek times, when Aesculapius, challenged with caring for a young Spartan girl with dropsy (edema), resorted to “[cutting] off her head, [turning] her upside down until the fluid ran out, and then [replacing] her head.” Other methods of mechanical fluid removal using leeches, bloodletting, and lancing remained popular for the treatment of edema until the 19th century, when an array of diuretic options emerged.

Infographic by @JMTeakell adapted from Eknoyan
We are now fortunate to have a number of diuretic (and natriuretic) agents in our armamentarium. Despite this, there remain cases where diuresis may be particularly difficult to achieve, tagged as “diuretic resistance.” Diuretic resistance may be an unrelenting force — sometimes difficult to diagnose and even harder to treat. In this section, the diagnosis and the treatment of diuretic resistance go head-to-head, and the winner likely has the tenacity it takes to go far in this year’s tournament.

Mechanisms of Diuretic Resistance
The concept of diuretic resistance is based on failure to achieve euvolemia despite increasing doses or maximal tolerated diuretic therapy. A diagnosis of diuretic resistance in patients can be challenging and involves both confirmation of volume status and evaluation of possible contributing factors. Congestion related to heart failure is one of the top reasons for hospital admission, and diuretic resistance leading to refractory congestion is associated with increased length of stay and other intermediate complications. Determining what is driving refractory congestion in a given patient is an essential first step in developing an effective treatment strategy. In other words, to beat the defense, we have to understand their game plan first.

Copyright: BLK Studio / Shutterstock
Mechanisms of Impaired Diuretic Response
Loop diuretics do their magic by blocking a major site of sodium reabsorption, the Na-K-2Cl co-transporter (NKCC2) in the thick ascending loop of Henle and macula densa. Sodium is the currency of the body — where sodium goes, water will follow — so diuresis occurs as a consequence of natriuresis. A normal physiologic diuretic response to 40 mg intravenous (IV) furosemide is 3-4 liters (L) of urine output containing 200-300 mmol of sodium in 3-4 hours. There are two main mechanisms of impaired diuretic response in heart failure: (1) insufficient delivery of loop diuretics to the proximal tubule, and (2) reabsorption of sodium at other nephron segments due to heightened sodium avidity.
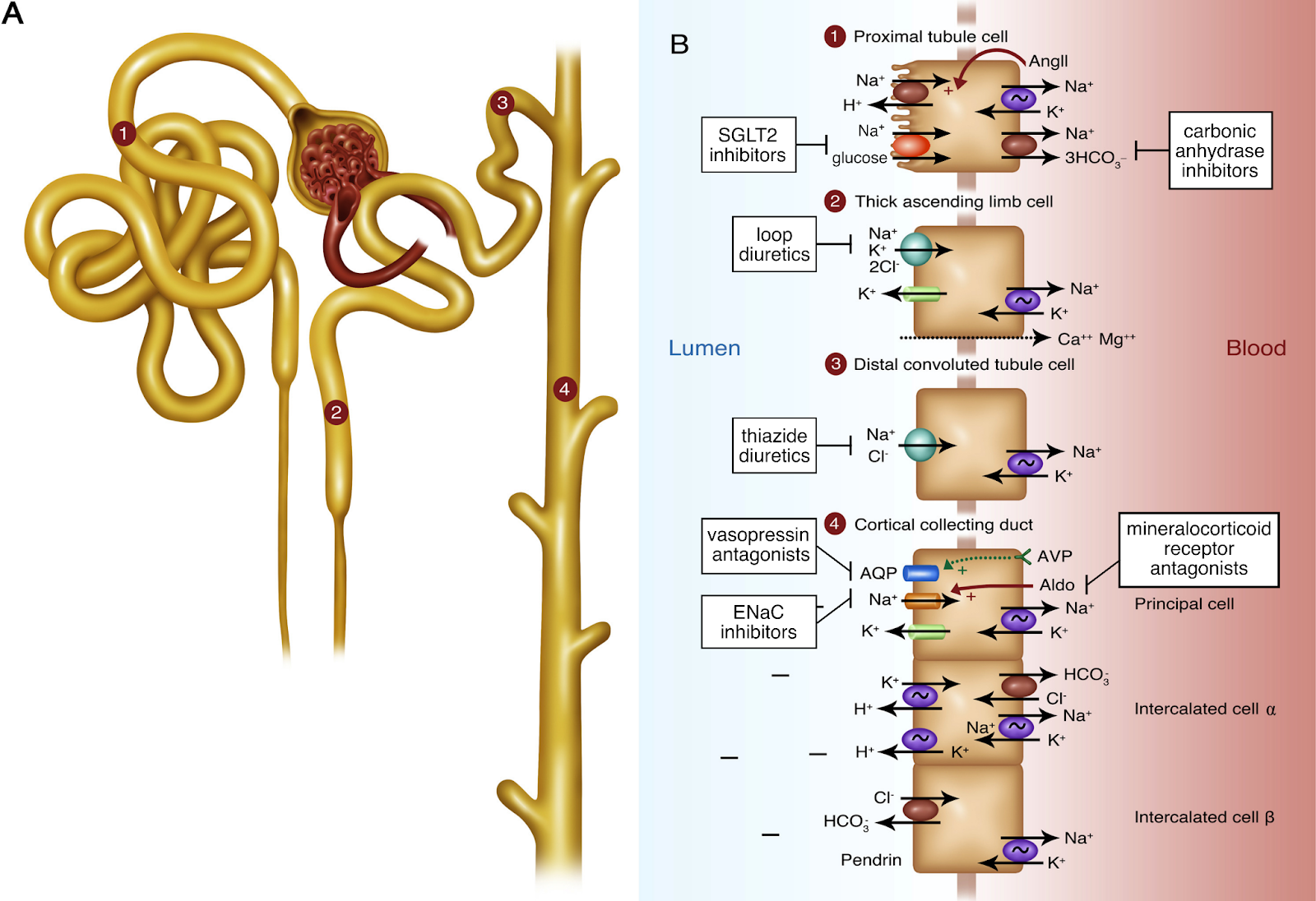
Sites of action of diuretics. Figure 1 from Danziger and Hoenig, AJKD, © National Kidney Foundation.
(1) Insufficient delivery of loop diuretics to the proximal tubule
A loop diuretic’s journey to its site of action in the kidney is long and fraught with potential hurdles. To review: orally administered loop diuretics are first absorbed in the stomach and small intestine. They then circulate in a highly protein-bound state, with 95%-98% normally bound to albumin, rendering them unable to be filtered by the glomerulus. So, the loop diuretic’s game plan is to get into proximal tubular epithelial cells from the peritubular capillaries via organic anion transporter (OAT) channels, and then ultimately to be secreted into the tubular lumen via multidrug resistance protein 4 (MRP4) channels. Finally, the intratubular diuretics make their way to the thick ascending limb, where their concentration determines the natriuretic effect.
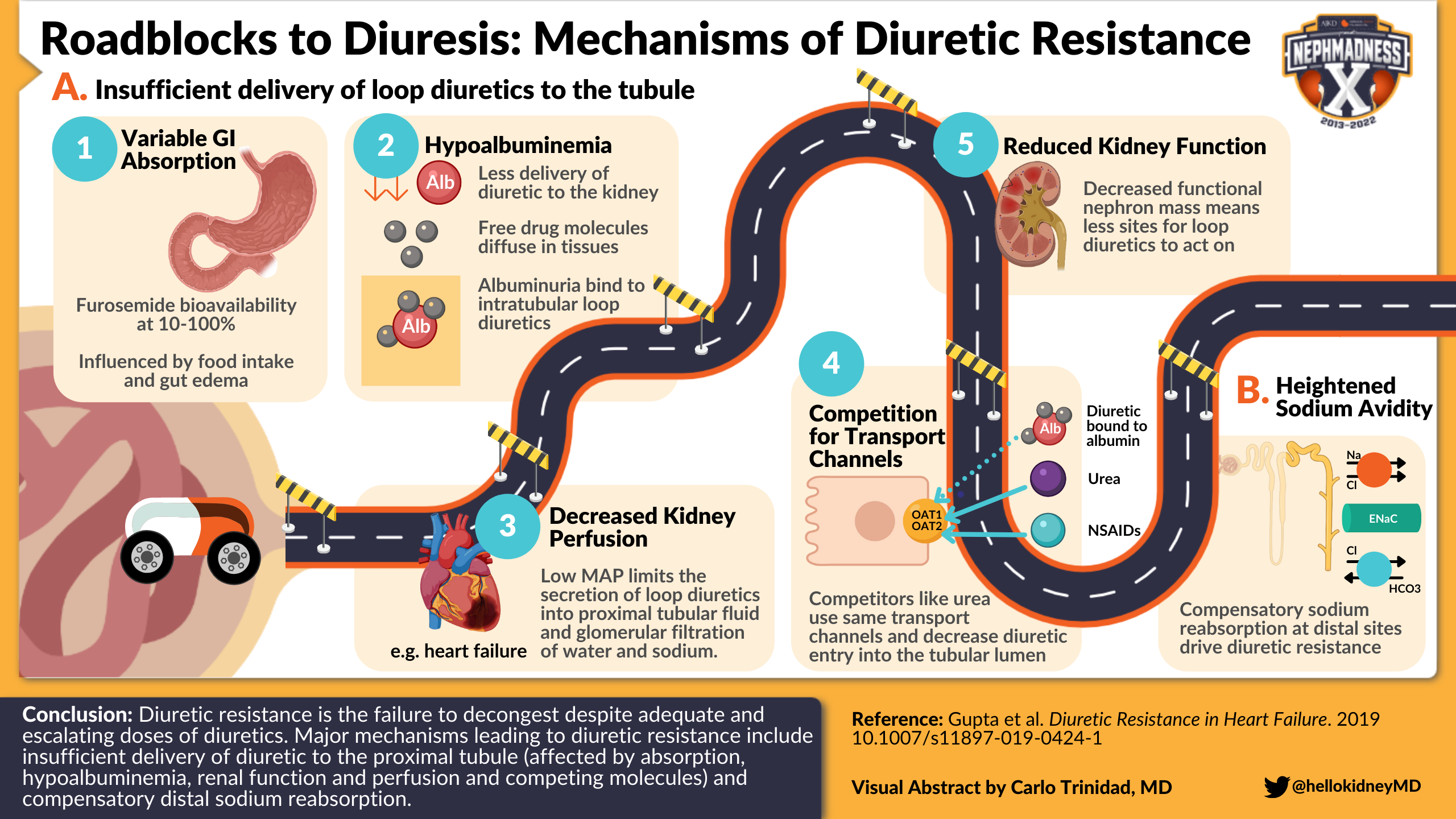
Visual abstract by @hellokidneyMD on Gupta et al
Let’s take a look at potential roadblocks during this journey that may culminate in decreased loop diuretic concentration at its site of action and impaired diuretic response. These include:
- Variable gastrointestinal absorption: Gastrointestinal absorption of furosemide is highly variable, with bioavailability ranging between 10%-100% (on average, 50%). There is significant variability between persons that is influenced by factors such as food intake and gut edema (or perhaps delayed gastric emptying). Oral torsemide and bumetanide have a more consistent and better bioavailability range (>80%).
- Hypoalbuminemia: Reduced availability of the albumin shuttle implies that less of the administered diuretic can be delivered to the kidney. Loop diuretics without a carrier are, in theory, free to diffuse and occupy a large volume of distribution. Conversely, albuminuria may bind to intratubular loop diuretics and reduce their efficiency within the loop of Henle. However, in a study of patients with a range of serum albumin levels (2.4–4.9 g/dL) and various degrees of albuminuria (median uACR 47 mg/g, IQR 17–305), serum albumin was not associated with diuretic delivery and albuminuria did not affect diuretic efficiency. However, it is unclear whether this is true at the extremes of hypoalbuminemia and heavy albuminuria, such as in nephrotic syndrome.
- Decreased kidney perfusion: Renal perfusion pressure is estimated as mean arterial pressure (MAP) minus central venous pressure (CVP). In other words, low MAP or high CVP can limit kidney perfusion. Decreased kidney perfusion can limit both the secretion of loop diuretics into proximal tubular fluid and glomerular filtration of water and sodium. One example is low cardiac output, a manifestation of advanced heart failure that causes multi-organ hypoperfusion and contributes to reduced diuretic efficiency, and in which plasma diuretics have a harder time reaching the kidney. While clinical history and exam (or improvement with a trial of empiric dobutamine) may suggest a low-output state, invasive hemodynamic assessment can more definitively assess cardiac output. Right heart catheterization is useful in the diagnostic workup of select patients with heart failure and reduced diuretic response. Check out episode 153 of the CardioNerds Case Report podcast series for a discussion of a case where obtaining a right heart catheterization was helpful.
- Reduced kidney function: Both chronic kidney disease (CKD) and acute kidney injury (AKI) can limit diuretic effectiveness for a conceptually simple reason; loop diuretics need a working target to do their thing. In the setting of reduced functional nephron mass, it is harder for loop diuretics to engage loops of Henle because there are simply less of them that still function properly. Thus, unsurprisingly, the maximal response to a diuretic decreases as CKD progresses and as AKI worsens. In fact, poor urine output in response to a “furosemide stress test” is one the best predictors of AKI severity. That being said, loop diuretics can be effective at all stages of CKD and in many cases of AKI, though higher doses are typically required.
- Competition at proximal tubule channels: Loop diuretics use the same turnstile to enter the proximal tubule lumen as other organic anions such as non-steroidal anti-inflammatory drugs (NSAIDs), β-lactams, methotrexate, urate, and urea — the OAT channel. Increased levels of these competitors can decrease diuretic entry into the tubular lumen and consequently limit their concentration in the loop of Henle. NSAID use in patients with heart failure has been associated with a 2-fold increased risk of hospitalization in an observational study. Of course, NSAIDs also reduce kidney perfusion; their effects on diuretic efficiency are thus manifold.
In each of these scenarios, there is incomplete blockade of NKCC2 channels because the needed dose of loop diuretic does not complete its journey to the loop of Henle. Some do not consider this “true” diuretic resistance, since if these problems with drug delivery were fixed and NKCC2 were fully blocked, diuresis would improve. In practice, however, these factors are hard to separate from —and often overlap with — the mechanism described in the next section.
(2) Heightened sodium avidity
Loop diuretics close the door to sodium re-entry at the level of the loop of Henle. Filtered sodium (ever the optimist) knows that when one door closes, another one (or three) may open. Distal to the NKCC2 transporter, there are Na-Cl symporters in the distal convoluted tubule as well as epithelial sodium channels (ENaC) and Na-dependent Cl/HCO3 exchangers (NDCBE) in the collecting ducts eager to assist sodium with its plan for re-entry. Compensatory sodium reabsorption at these distal sites is a main driver of diuretic resistance in patients with heart failure. Let’s explore what drives sodium avidity in heart failure.
Several human studies in the 1980s formed the basis of our understanding of how our kidneys respond to loop diuretics. Administering a loop diuretic to several cohorts of euvolemic, healthy volunteers provided roughly 6 hours of natriuresis (with high urine sodium), followed by a period of compensatory sodium reabsorption (with low urine sodium). This is known as the “braking phenomenon”, and is a protective mechanism triggered by acute sodium and water loss to avoid over-diuresis. It has become conventional wisdom to extrapolate this concept to heart failure; dips in serum sodium and blood volume with each dose of loop diuretic would activate the renin-angiotensin system (RAS) and trigger sodium reabsorption once the diuretic had worn off.
However, there was a flaw in this long-held theory: IV diuretic infusions, which obliviated the post-diuretic “braking” period, did not fix the sodium reabsorption problem in advanced heart failure. More recently, a landmark mechanistic paper established that compensatory post-diuretic renal sodium reabsorption is not a dominant mechanism of diuretic resistance in acute heart failure. Rather, patients with advanced heart failure and diuretic resistance seem to be in a continuous sodium-avid state.

Visual abstract by @sophia_kidney on Cox et al.
Neurohormonal adaptations and structural changes in the distal nephron are thought to underlie this steady sodium avidity. RAS overactivation is common in advanced heart failure with diuretic resistance, despite standard-of-care RAS blockade with angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs). The effects of “aldosterone escape” in heart failure include direct activation of the epithelial sodium channel (ENaC) and NDCBE-pendrin system in the collecting ducts and hypertrophy of the distal convoluted tubule, which increases the number of apical Na-Cl symporters available for sodium reabsorption. In other words, chronic RAS activation seems to be the doorstop that holds NKCC2, ENaC, and NDCBE open, ultimately leading to diuretic resistance in chronic heart failure.

Left panels: Photomicrographs of rat kidneys after administration of furosemide showing hypertrophy of distal convoluted tubule (DCT) cells, connecting tubule (CN) cells, principal cells, and intercalated cells. Right panels: Transmission electron micrographs of DCT cells from rats administered furosemide showing taller cells, with increased height of lateral cell processes and larger more rounded nuclei. Adapted from Ellison et al. © Journal of Clinical Investigation, used with permission.
Distinguishing Between the 2 Main Causes of Impaired Diuretic Response
Diagnosing diuretic resistance and identifying the driving cause is difficult in practice. A spot urine sodium collected two hours after administering a diuretic can help predict whether or not natriuresis will be adequate and can facilitate early recognition of an impaired diuretic response. However, determining whether this is due to insufficient loop diuretic delivery or distal sodium reabsorption is more challenging. A method used in research studies is to measure the relative changes in the fractional excretion of sodium (FeNa) and endogenous lithium (FeLi). This relies on the premise that while both cations are reabsorbed via NKCC2, lithium is not reabsorbed in the distal nephron, and so a disproportionate increase in FeLi after diuretic administration would suggest that distal sodium reabsorption is at play (see Figure 1 of this paper by Rao et al. for more). The ongoing Mechanisms of Diuretic Resistance Study should help elucidate how best to discriminate etiologies of diuretic resistance.
Assessing Volume Status
When thinking about diuretic resistance, we must also consider a different possibility: does the patient truly have elevated left-sided ventricular filling pressures leading to refractory systemic congestion? The volume status exam aims to estimate total body water distribution and related hemodynamics. We assess manifestations of right-sided filling pressures (eg, jugular venous pressure height, peripheral edema grade) as surrogates of left-sided ventricular filling pressures. Serial changes in exam findings, patient weight, and fluid output are commonly used to monitor response to diuresis, as recommended by major society guidelines. However, reliable metrics can be difficult to obtain and, even when available, they can be discordant with true fluid status.
There are newer adjunct modalities that can help us more precisely assess volume status. Bedside ultrasound can be used to confirm jugular venous distension, organ congestion (eg, pulmonary edema), and third-spacing of fluid (eg, pleural effusions and ascites). More recently, the Venous Excess Ultrasound Score (VExUS) has emerged as a popular tool to assess venous congestion. VExUS is a scoring system developed in 2019 to identify and grade systemic congestion severity based on inferior vena cava diameter and flow patterns in the hepatic, portal, and renal veins. For more on VExUS, check out this video, or this Renal Fellow Network post.

VExUS Score Pocket Card from POCUS 101, used with permission.
Other adjuncts that can help assess total body water distribution include bioimpedance and radio-labeled albumin dilution. However, these require specialized equipment to implement. Ultimately, estimation of filling pressures with right heart catheterization may be required for certain challenging cases to confirm that there is actually ongoing hypervolemia that requires decongestion.
Treatments of Diuretic Resistance
Depending on the diuretic resistance driver, one of several emerging strategies or “trick plays” may be useful in overcoming the hurdles that may impede effective diuresis. This section will discuss these treatments as well as some safety considerations when employing them.

Copyright: Alohaflaminggo / Shutterstock
A. Maximizing Loop Diuretic Efficacy
Changing the route of loop diuretic administration (IV vs oral) or increasing the dose/frequency may be required in patients who are suspected to have reduced diuretic efficiency.
Loop diuretics have an S-shaped dose-response curve:

Dose response curve of loop diuretics. Adapted from Braunwald.
The “ceiling dose” refers to the single dose amount that is required to reach the ceiling of diuretic efficiency; doses above this ceiling do not meaningfully increase diuresis. While 80 mg of oral furosemide is a typical ceiling dose for “diuretic-naïve” individuals, increased doses of oral diuretics can be required in the setting of impaired kidney drug delivery. For example, in CKD, ceiling doses can be as high as 400 mg of oral furosemide per dose. Once the diuretic ceiling dose has been identified for an individual, this dose can be given up to every 6 hours to maximize the total daily urine output. The ceiling dose can change in an individual over time, pending the factors that impact loop diuretic drug delivery and efficacy discussed in the previous section.
Furosemide is the most prescribed loop diuretic in North America, largely because it has historically been cheaper than other diuretics. Torsemide and bumetanide are becoming increasingly popular first line choices due to improving costs and beneficial clinical profiles. They have more reliable bioavailability (80%-100%; compared to 50% for furosemide), and can be more effective than oral furosemide when gut edema is present. Bumetanide is the most potent of the three: 40 mg furosemide PO is equivalent to 1 mg bumetanide PO/IV or 20 mg torsemide PO/IV. Torsemide has the longest duration of action, and has additionally been associated with reduced cardiac fibrosis in small trials. A randomized trial comparing the mortality benefits of torsemide versus furosemide is underway.
For safe use, it is important to keep in mind that furosemide metabolism is affected by kidney function. Furosemide is mainly eliminated unchanged in the urine, but it is also metabolized to its inactive form in the kidney parenchyma. Bumetanide and torsemide are conversely metabolized in the liver. So, in advanced CKD, serum levels of furosemide can rise more easily, putting patients at higher risk for ototoxicity. Ototoxicity associated with loop diuretics is often — but not always — reversible.
IV diuretics are often used in the inpatient setting to ensure the ceiling dose is attained. A recommended diuretic dose for patients admitted to hospital is 2.5x their home dose given intravenously (ie, 40 mg PO at home → 100 mg IV recommended initial dose). Measurement of a spot urine sodium 1-2 hours after diuretic administration can be used to assess whether the current dose of diuretic is adequate (the goal is a UNa > 50 mmol/L), and this value can be input into a validated Natriuretic Response Prediction Equation to adjust subsequent diuretic doses. If the response to the initial dose is suboptimal, a subsequent higher dose can be given immediately in order to minimize the time-to-decongestion.
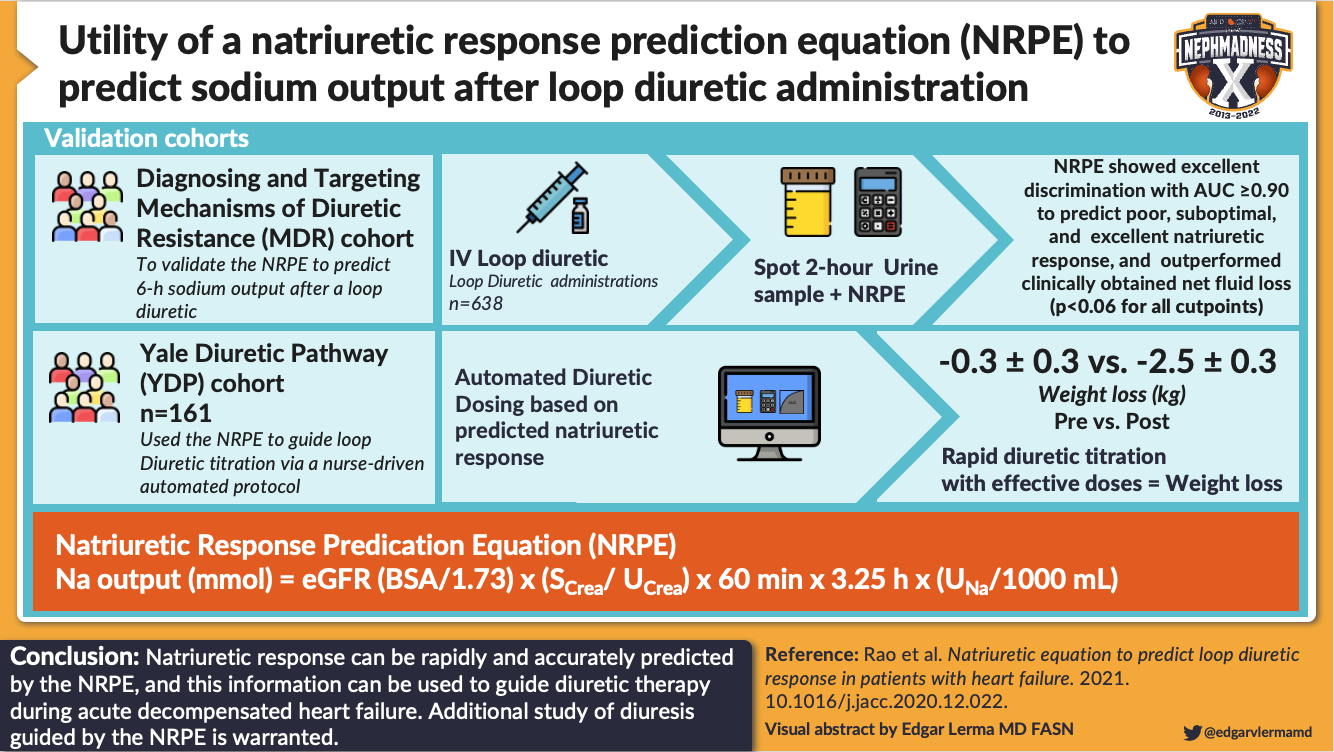
Visual abstract by @edgarvlermamd on Rao et al.
What about continuous IV loop diuretic infusions? In the DOSE trial (n = 308), continuous loop diuretic infusions (CLDI) were not more effective than interval bolus dosing for acute decompensated heart failure, though a subsequent meta-analysis of 8 randomized control trials (RCTs) studying this question (n = 669) showed increased urine output and weight loss for patients receiving CLDI. Retrospective analysis of 5,738 participants in the ASCEND-HF trial revealed that patients receiving CLDI received a total higher cumulative dose of diuretics (a median of 160 mg for intermittent dosing vs 264 mg for continuous dosing) and output about half a liter more urine on average per day. However, urine output was not significantly greater when adjusted for diuretic dose. Since diuretic route and dosing schedule was left to the discretion of the treating physician in ASCEND-HF, the study sheds some light on real-life practice trends; patients who received diuretic infusions tended to be sicker. When large doses are required, it seems physicians and nurses felt more comfortable with continuous delivery of a lower dose, to conceptually avoid high serum peaks and related potential ototoxicity. However, we don’t have great evidence that this actually makes a difference.
Other than maximizing the diuretic dose, what else can we do? Here are a few adjuvants that have been tried to help mitigate barriers to diuretic efficiency:
- Albumin infusions were not consistently effective at augmenting diuresis in diuretic resistance with hypoalbuminemia across trials assessing this strategy, though they may be beneficial in select cases of profound hypoalbuminemia. Sulfisoxazole, an agent that prevents albumin-loop diuretic binding in the urine, is also not an effective adjuvant in nephrotic syndrome.
- In select situations where kidney perfusion is compromised, medications used to improve organ perfusion can improve diuresis by increasing glomerular filtration rate and diuretic delivery to the kidney. For example, in patients with low cardiac output, inotropes such as dobutamine and milrinone can improve kidney perfusion. In patients with chronically low systemic vascular resistance (SVR) due to comorbidities such as cirrhosis, therapies raising SVR such as octreotide and midodrine can improve kidney perfusion. Other drugs that can improve kidney perfusion include low-dose dopamine and synthetic natriuretic peptides (eg, nesiritide), which promote kidney arterial vasodilation. Unfortunately, these treatments are not broadly effective at improving decongestion in patients with acute heart failure and CKD stages III-IV, as shown in the ROSE-AHF trial. However, there may be a role for low-dose dopamine in select settings. The role of renal negative pressure treatment is under study in animal models.
B. Sequential Nephron Blockade to Prevent Distal Sodium Reabsorption
Thiazides
A key strategy that is used to prevent distal sodium reabsorption — the major driver of diuretic resistance — is blocking these ports of re-entry. Thiazide and thiazide-like diuretics block the Na-Cl symporter in the distal convoluted tubule and the NDCBE-pendrin system in the collecting ducts. Combination treatment with a loop diuretic and thiazide diuretic (eg, metolazone) is often deployed to treat diuretic resistance, and conventionally referred to as “sequential nephron blockade”. This approach was first formally studied in 1996. There is a major safety point to be aware of with this strategy: both agents promote natriuresis and kaliuresis, and concomitant use can result in drastic lowering of serum sodium and potassium. These electrolytes must be monitored frequently (metolazone has a long duration of action of 12-24 hours), and potassium repletion may be required in patients being treated with sequential nephron blockade.
Mineralocorticoid Receptor Antagonists
Mineralocorticoid receptor antagonists blunt sodium avidity in two ways: they antagonize ENaC and therefore block distal sodium reabsorption, and they also inhibit RAS activation. While many patients with advanced heart failure with reduced ejection fraction will be on low-dose spironolactone (12.5-25 mg) as part of guideline-directed medical therapy, high-dose spironolactone has been hypothesized to provide additional benefit through greater RAS blockade (analogous to how it is used in volume management in decompensated cirrhosis). However, in a large multicenter RCT of 100 mg vs 25 mg spironolactone in patients with heart failure (ATHENA-HF), decongestion was not meaningfully improved with the higher dose. Experts suspect that incomplete RAS blockade was at play in ATHENA-HF, and that high-dose spironolactone may confer benefit in select populations, such as those with evidence of diuretic resistance or high measured serum aldosterone levels.
Acetazolamide
Acetazolamide is the oldest “modern diuretic” in use. It replaced post-war mercurial agents and was the most commonly used diuretic agent for decongestion in heart failure in the 1950s. Acetazolamide is a carbonic anhydrase inhibitor that blocks sodium re-entry in the proximal tubule and in the collecting ducts (it blocks the NDCBE-pendrin system). While it is a fairly poor diuretic on its own, its use in combination with other diuretics has been the subject of investigation in recent years. In a small RCT (n = 34), acetazolamide increased loop diuretic efficiency in patients with diuretic resistance, and a larger RCT (the ADVOR trial) is ongoing. Another small trial (n = 20) showed that the addition of acetazolamide to a loop and thiazide diuretic regimen also augmented diuresis.
A combination regimen including all four of the above-mentioned medications — a loop diuretic, thiazide, acetazolamide, and spironolactone — is called multi-nephron segment diuretic therapy. Multi-nephron segment diuretic therapy appears to be effective in treating diuretic resistance without significant electrolyte abnormalities based on retrospective analysis of 167 cases.
Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors
The newest class of sodium channel blockers with a proven role in augmenting diuresis are the sodium-glucose cotransporter 2 (SGLT2) inhibitors. As their name implies, they block sodium and glucose reuptake via the SGLT2 channel in the proximal tubule. A mechanistic study examining the diuretic effects of empagliflozin in human subjects reported modest natriuresis when added to loop diuretics (check out NephJC for more). The increased distal sodium delivery activates tubuloglomerular feedback (remember #NephMadness 2020?), counteracting the pathologic inhibition that occurs with loop diuretics in heart failure. SGLT2 inhibitors reduce the risk of hospitalizations and death in patients with heart failure, including those without diabetes and those with preserved ejection fraction. In the future, we may see polypills that combine SGLT2 inhibitors with other diuretics for patients with heart failure.
C. “Hyperdiuresis” & Chloride Repletion
“Hyperdiuresis” refers to a relatively recent strategy of giving a small bolus of hypertonic saline (eg, 150 mL of 3% NaCl administered over 30 minutes) with a loop diuretic, a strategy seemingly at odds with our dogmatic reflex to advise sodium restriction in heart failure (despite inconsistent evidence to support this practice). Acute increases in serum sodium and chloride concentrations may augment diuresis via several mechanisms, including drawing out interstitial fluid by osmosis and blunting RAS activity. Hyperdiuresis has been shown to be effective in several small trials and one large RCT (reviewed in NephMadness 2015, with additional supporting evidence in a more recent retrospective study covered here). This approach should be employed cautiously, given the possibility of acutely worsened volume overload, rapid sodium elevation, and other electrolyte abnormalities.
As hypochloremia is common in heart failure and is associated with low diuretic efficiency, chloride replenishment may explain the success of this strategy. On the tail of promising pilot studies with oral lysine chloride, larger trials are ongoing to determine if oral chloride replacement can effectively reduce blood volume in patients with diuretic resistance.
D. Vasopressin Antagonism
Vasopressin — also known as antidiuretic hormone — is elevated as part of pathologic neurohormonal activation in heart failure that drives sodium avidity. It makes sense to block the antidiuretic hormone if we want diuresis … right? Vasopressin acts on the collecting ducts of the kidney to reabsorb free water, decreasing plasma sodium chloride concentrations, which promotes salt reabsorption. Based on this premise, the vasopressin receptor blocker tolvaptan was put to the test in EVEREST, a study of short- and long-term outcomes in heart failure. While tolvaptan mediated greater urine output in the short term, this did not translate to long-term clinical benefits. In subsequent studies (Felker 2017 and Konstam 2017), patients did not report improved short-term dyspnea with the addition of tolvaptan, despite greater urine output.
E. Ultrafiltration (UF)
Unfortunately, there are situations in which all the diuretics in the world won’t effectively mobilize fluid. UF is the removal of isotonic fluid via convection, typically achieved using a dialysis machine. It is often considered in patients who have acute congestion refractory to multiple diuretics strategies, or those who have severe AKI or advanced CKD limiting diuretic efficacy. UF can be a short-term strategy used to support volume removal pending kidney recovery. However, there is limited data supporting its widespread use. In CARRESS-HF, UF was inferior to a stepped pharmacologic treatment strategy. UF has been associated with harm; those who were randomized to receive UF in CARRESS-HF and in the AVOID-HF trial experienced significantly more adverse effects than those on pharmacologic treatment.
Finally, diuretic resistance may be unavoidable and even unsurmountable as part of the natural course of end-stage heart failure. If patients develop persistent New York Heart Association (NYHA) class III/IV symptoms with waning treatment options, involvement of palliative care colleagues can assist patients with the physical symptoms and psychosocial sequelae.
Check out this podcast episode of The Curbsiders featuring Sadiya Khan @HeartDocSadiya and Joel Topf @kidney_boy:
#326 Cardiorenal Syndrome
COMMENTARY BY ERIN MICHOS:
The Year That Cardiology and Nephrology Are Friends
Cardiac Biomarkers in CKD vs Kidney Biomarkers in CHF
Cardiac Biomarkers in CKD
Biomarkers are indicators of a biological state that can help guide clinical decision-making by conveying the silent tales of struggling organs. However, interpreting these biomarkers can be difficult when more than one organ is struggling. Do a team’s stats always tell the whole story? This region will discuss cardiac biomarkers in patients with kidney disease, and kidney function markers in those with heart disease.

Marker drawing by Meera Sparks. Reproduced with permission.
Cardiac Biomarkers in Patients with Kidney Disease
Cardiac biomarkers are important in the diagnosis of cardiovascular disease (CVD), but interpretation may not be straightforward in patients with impaired kidney function. CVD is the leading cause of death in patients with CKD and about 70% of patients with CKD have concomitant CVD. However, CVD can be difficult to recognize in those with CKD or ESKD, in part due to challenges related to the interpretation of cardiac biomarkers. Cardiac biomarkers are considered signposts of underlying cardiac injury or strain in the general population: elevated troponin may suggest myocardial ischemia or acute coronary syndrome (ACS), and elevated brain natriuretic peptide (BNP) signals volume overload in decompensated heart failure. However, both troponin and BNP are chronically elevated in at least 40% of individuals with CKD without clinical CVD. Why does this happen, and is it associated with cardiac risk?
Troponins
Troponin is a protein complex formed of 3 subunits — troponin C, troponin I, and troponin T — that facilitates contraction of cardiomyocytes and skeletal muscle myocytes (Figure 8A). When a muscle is injured, troponin is released in the bloodstream. Cardiac-specific isoforms of troponin I (cTnI) and Troponin T (cTnT) are measured clinically to detect myocardial injury including, but not limited to, ACS. Shortly after its introduction as a cardiac biomarker in 1990, cTnT was recognized to be chronically elevated in patients receiving dialysis (later also reported across CKD stages). While this was initially considered to be a false positive assay artifact, it was confirmed in 1998 that the chronic cTnT elevations observed CKD were, in fact, cardiac in origin. Intact cTnT and cTnI are too big to be filtered by the glomerulus, weighing 37 and 29 kDa, respectively. Degraded troponin fragments smaller than 20 kDa in size can be filtered and released in the urine; however, a recent mechanistic study of cTnT clearance showed that only about 8%-19% of total cTnT is excreted by healthy kidneys in the steady state. Newer high-sensitivity (hs) cTnI and cTnT assays have been recently introduced into clinical practice to enable early detection of myocardial infarction (MI). Non-specific troponin elevations in CKD are also observed using these high sensitivity assays, but are not without risk.

Cardiac biomarkers and kidney filtration. A) Structure of the troponin complex. Troponin I and troponin T are measured clinically. B) ProBNP is cleaved into N-terminal proBNP (NT-proBNP) and BNP. Both are measured clinically. C) NT-proBNP, BNP, and troponin degradation products are filtered through the glomerulus. Illustration by Caitlyn Vlasschaert, created with BioRender.com.
A recent meta-analysis estimated that, in patients on dialysis, chronically elevated troponin is associated with a three-to-four times greater risk of CVD-related death (HR 3.3 for cTnT, 95% CI: 1.8–5.4; HR 4.2 for cTnI, 95% CI: 2.0–9.2). Risk across broader estimated glomerular filtration rate (eGFR) strata has been extensively studied more recently. In the Chronic Renal Insufficiency Cohort (CRIC), a prospective observational study of over 3,000 individuals with CKD, elevations in cTnT have been independently associated with increased risks of incident heart failure, incident atrial fibrillation, and incident CKD progression, as well as all-cause and CVD-related mortality. Similar findings are reported in the Atherosclerosis Risk in Community (ARIC) study and in the African American Study of Kidney Disease and Hypertension (AASK) study. Moreover, increased cTnT has been associated with left ventricular hypertrophy (LVH) in CRIC, and hs-cTnT has been associated with more subtle alterations of left ventricular structural alterations and incident LVH in the KNOW-CKD cohort.

Visual abstract by @Errantnephron on Bansal et al, Lamprea-Montealegre et al, Bansal et al, and Wang et al
A diagnosis of acute myocardial infarction (AMI) requires signs and symptoms of ACS: chest pain, detection of an elevated troponin (above the 99th percentile of a normal reference population), and evidence of myocardial ischemia. The most common type of ACS in individuals with CKD is a non-ST elevation MI (NSTEMI). Strikingly, less than 50% of patients with CKD or ESKD who present with AMI report classic ischemic symptoms of chest, arm, shoulder, or neck pain; anginal equivalents such as shortness of breath are more common. In a systematic review commissioned by Agency for Healthcare Research and Quality, troponin T (TnT) assays using the same upper limit as in the general population were found to remain sensitive in the CKD population (sensitivity 71%-100%) whereas troponin I (TnI) assays were only 43%-94% sensitive. Using standard upper reference limits, the specificity for a diagnosis of ACS was modest (31%-86% for TnT and 48%-100% for TnI). High sensitivity cardiac-specific isoform (cTn) assays are similarly sensitive enough to readily rule out AMI but lack the specificity required to diagnose AMI as easily as in the general population. A recent cohort analysis of the Canadian study of PREDiction of Death, Dialysis, and Interim Cardiovascular evenTs (CanPREDDICT) showed that using eGFR-specific cutoffs could improve reclassification in patients with advanced CKD. Reviewing trends in troponin change — including serial values obtained during a suspected ACS event and in comparison to a baseline “healthy” troponin value — have been suggested to facilitate ACS diagnosis, particularly in patients with CKD.
Natriuretic Peptides
BNP is a peptide hormone produced mainly by cardiomyocytes in response to cardiac wall stretch. The BNP pro-peptide is cleaved into N-terminal proBNP (NT-proBNP) and the active BNP hormone upon secretion into the bloodstream (Figure 8B). The kidney is the main site of action of active BNP; it increases GFR via renal vasculature vasodilatation and inhibits renin release, leading to enhanced diuresis and natriuresis. BNP and NT-proBNP are used clinically to assess whether decompensated heart failure is present, with specific thresholds based on age and sex in the general population. While BNP levels are still useful to help differentiate the cause of dyspnea in patients with CKD not on dialysis, higher thresholds have been suggested. Of note, BNP and NT-proBNP levels are lower in patients with obesity.
Both BNP and NT-pro-BNP are commonly chronically elevated in kidney disease; about 50% of patients with CKD and almost all dialysis patients have elevated values. BNP and NT-proBNP are both small (8.5 and 3.5 kDa, respectively) and thus freely filtered at the glomerulus. While BNP can be cleared in other ways (such as neprilysin-mediated cleavage), the kidney fractional excretion of BNP and NT-proBNP are similar at ~20% irrespective of kidney function. This suggests that loss of GFR alone would not lead to meaningful rises in serum troponin or BNP, and that chronic elevations observed in CKD are likely representative of chronic cardiac stress. Similar to troponin, chronic elevations in BNP portend poor clinical outcomes in CKD, including higher rates of incident heart failure, incident atrial fibrillation, incident CKD progression, and death. In the future, chronic troponin and BNP levels may be used to improve risk stratification for these adverse events, but large prospective studies are needed to assess their utility in this regard.
Kidney Biomarkers in Patients with Cardiac Disease

Marker Drawing by Meera Sparks. Reproduced with permission.
Titration of medical therapies in heart failure, including RAS inhibitors, SGLT2 inhibitors, and diuretics, may result in rises in serum creatinine. Clinicians concerned about kidney function may dose-reduce or stop guideline-directed medical therapy—but is it really kidney injury? We expect, and thus may accept a certain degree of creatinine elevation based on the pharmacology of these medications, a concept called “permissive hypercreatininemia” (ie, being OK with the rise in creatinine). Why should we permit hypercreatininemia and how do we know if the kidney is actually injured by these “nephrotoxins”?
Let’s start with diuretics, a common arena of debate. Should we start or stop them? Increase or decrease?
https://twitter.com/DGlaucomflecken/status/1373475652687388674
Rises in creatinine after initiation of or increases in diuretic dosing can be a challenge. Is it a sign that kidney function is being affected? Some practitioners incorporate creatinine fluctuations in their holistic assessment of dynamic volume status change to guide further titration — but is this correct? Should a rise in creatinine prompt us to think twice about pushing diuresis? Incomplete decongestion of patients with heart failure in the setting of rising creatinine happens more often than we may realize; one-fifth of patients in the ADHERE registry were discharged after an admission for heart failure without weight loss, which is associated with worse outcomes. Indeed, in retrospective analyses of the ESCAPE and DOSE trials, hemoconcentration and creatinine rises meeting AKI criteria were paradoxically associated with reduced morbidity and mortality.
In the ROSE-AHF trial, changes in kidney filtration markers (creatinine, cystatin) after aggressive diuresis were not correlated with changes in kidney injury biomarkers (neutrophil gelatinase-associated lipocalin [NGAL], N-acetyl-β-D-glucosaminidase [NAG], and kidney injury molecule 1 [KIM-1]). Injury biomarkers were also not elevated in the group of patients with creatinine elevations compared to in those without creatinine elevations. Altogether, this suggests that we should prioritize clinical evidence of ongoing hypervolemia over changes in creatinine in diuresis, but that can be easier said than done. Scared of diuresing too quickly? A recent study found that rapid decongestion of patients with acute heart failure did not worsen kidney outcomes.
Dr. Janani Rangaswami, lead author of the 2019 Scientific Statement from the American Heart Association on Cardiorenal syndrome, has shared that with particularly challenging cases she sometimes checks a spot urine NGAL to screen for acute tubular injury so that she can push forward with diuretic therapy with greater confidence. In a similar vein, urinalysis can be used to help to look for acute tubular necrosis in various clinical settings (as reviewed in NephMadness 2021). Another option is the NephroCheck panel, available in the US, which measures 2 other biomarkers of acute kidney injury: TIMP-2 (Tissue Inhibitor of Metalloproteinase 2) and IGFBP-7 (Insulin-like Growth Factor Binding Protein 7) proteins. Unfortunately, none these strategies have been tested systematically in heart failure, and identifying reliable methods to assess whether hypercreatininemia signals AKI has been identified as a key gap in knowledge in heart failure treatment.
Let’s move onto a couple other creatinine-raising “nephrotoxins”: SGLT2 inhibitors and RAS inhibitors, such as ACE inhibitors and ARBs.
Both RAS inhibitors and SGLT2 inhibitors reduce intraglomerular pressure. RAS inhibitors accomplish this by promoting efferent arteriole vasodilation, while SGLT2 inhibitors promote afferent arteriolar vasoconstriction (throwback to NephMadness 2020!). This reduces pressure within the glomerulus, which is nephroprotective; kidney function is better preserved over time, as shown in numerous trials. Indices of glomerular filtration such as creatinine and cystatin C commonly rise when initiating RAS inhibitors or SGLT2 inhibitors because reduced intraglomerular pressure means that these are less readily filtered into the tubules. It is important to be aware that these drugs are expected to reduce GFR; it is a sign that they are protecting the kidney.

Visual abstract by @hellokidneyMD on Meraz-Munoz et al.
However, concern arises when someone already on stable doses of ACE inhibitors, ARBs, and/or SGLT2 inhibitors develops an acute creatinine rise, or when they become ill and at risk of hypovolemia. When do we hold these medications, and when can they safely be restarted? There are no RCTs to help guide these clinical scenarios, and as a result, this practice is subject to high inter-prescriber variability. Most clinicians consider it “reasonable” — as we like to say — to hold ACEi/ARB and SGLT2 inhibitors during intercurrent illness. However, there is a paucity of data guiding this.
Here’s an example of guidance that may be given to patients on which medications to hold on a “sick day.” An overlooked issue is that patients are often not given clear guidance as to when to restart these medications. A large observational study covered recently in NephJC suggested that stopping an ACEi/ARB prescribed before hospital admission and not restarting it before discharge was associated with higher risk of death during the 2-to-7 year observation period (HR, 1.23; 95% CI, 1.17-1.30). However, this practice is shockingly common; in one multi-center study, more than 75% of patients had not resumed their ACEi or ARB by their first follow-up appointment after in-hospital AKI (about 1 month post-discharge).
To recap: hypercreatininemia should be permitted when titrating loop diuretics to achieve euvolemia (unless there is evidence of kidney injury) and is expected upon initiation of SGLT2 inhibitors and RAS inhibitors since they lower intraglomerular pressure. Though these medications are often held during intercurrent illness to prevent kidney injury, remembering to restart them is important. In other words, don’t keep your star player on the bench for the entire season if it’s safe for them to come back.
– Executive Team Members for this region: Samira Farouk @ssfarouk and Anna Burgner @anna_burgner
How to Claim CME and MOC
US-based physicians can earn 1.0 CME credit and 1.0 MOC per region through NKF PERC (detailed instructions here). The CME and MOC activity will expire on June 1, 2022.
Click to read more NephMadness 2022 Regions
Nephrocheck does not contain NGAL.The statement needs to be modified. Excellent post
Thank you for alerting us – we have corrected the sentence.